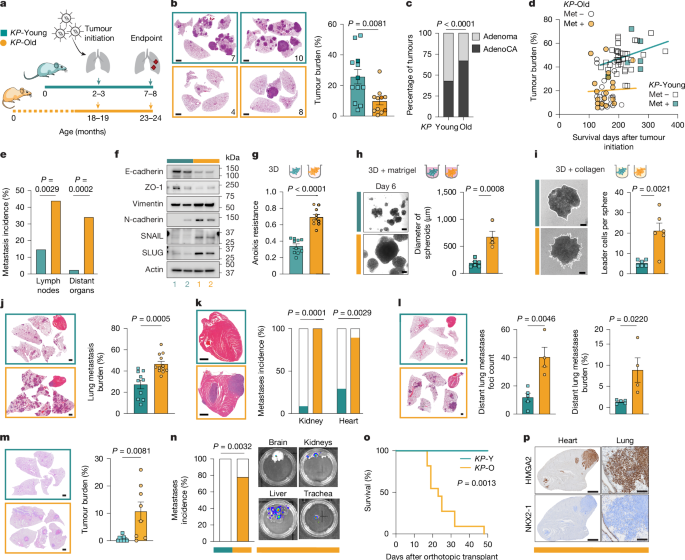
Ageing promotes metastasis via activation of the integrated stress response
Mice and in vivo studies
KrasLSL-G12D/+Trp53flox/flox mice (designated as KP)60 were maintained on a mixed C57BL/6-129/Sv genetic background. Lung tumours were induced in young KP (2–3 months old; referred to as KP-Young) and old KP (18–19 months old, referred to as KP-Old) mice through intratracheal instillation with Lenti-Cre as described in ref. 61 or with Ad5mSPC-Cre viral particles (PFU 2 × 107; University of Iowa; VVC-Berns-1168) under general anaesthesia as described in ref. 10.
NXG (NOD Xenograft Gamma) mice (NOD-Prkdcscid–IL2rgTm1/Rj) were obtained from Janvier Laboratories and were 6–10 weeks old at the start of the experiments. All transplantation studies were conducted using age-matched cohorts of mice. For subcutaneous implantations, a total of 2.5 × 105 of green fluorescent protein (GFP)-luciferase-expressing versions of the indicated cells suspended in PBS were subcutaneously injected into the lower right and lower left flanks of NXG mice. Tumours were measured every other day by callipers and the tumour volume was calculated using the formula volume = (length × width2)/2. At no point during the study did any tumour exceed the permitted maximal measurements of 2 cm × 2 cm or endpoint criteria.
For intravenous injections, a total of 5 × 104 of the indicated cells were injected into the lateral tail vein of NXG mice. All transplantation experiments were performed in young immunodeficient NXG hosts to minimize confounding by host age or immunity.
For CB-839 studies, mice were randomized and subjected to treatment with either 200 mg kg−1 body weight CB-839 (no. HY-12248, MedChemExpress) or vehicle. Treatments were administered twice a day every other day following either the tumour-establishment phase (subcutaneous implantations) or within 6 h after the bloodstream injection of cells. The vehicle control consisted of 25% (w/v) 2-hydroxypropyl-β-cyclodextrin in 10 mM citrate buffer (pH 2.0).
For doxycycline-induced Atf4 silencing, 1 mg ml−1 of doxycycline (no. HY-N0565B, MedChemExpress) was administered in the drinking water supplemented with 5% sucrose. For ISRIB (no. HY-12495, MedChemExpress) studies, indicated cells were pretreated 3 times a day for 7 days with either ISRIB (2 µM) or vehicle (dimethylsulfoxide, DMSO) and then injected into mice. ISRIB was administered at 10 mg kg−1 twice daily by intraperitoneal injection or vehicle alone (45% saline, 50% PEG 400, 5% DMSO).
Same-sex mice were housed in individually ventilated cages, under a 12–12 h light–dark cycle, with ambient temperature (15–21 °C) and humidity control (45–70% relative humidity), enrichment material and ab libitum rodent chow and water. All mouse experiments described in this study were approved by the Research Animal Ethics Committee in Gothenburg (2071/19; 2077/19 and 6057/24).
IVIS imaging
Mice were injected intraperitoneally with 30 mg ml−1 of d-luciferin (no. 15225733, Fisher Scientific) and organs were imaged ex vivo 10 min after injection. Luminescence was quantified as constant, or as total flux (p s−1). Analysis was performed with Living Image v.4.7.4 software, maintaining the region of interest over the tissues at a constant size. The radiance (p s−1 cm−2 sr−1) from the region of interest was normalized against the background radiance.
Histology and IHC analyses
Lungs were perfused through the trachea with PBS, fixed overnight, transferred to 70% ethanol, embedded in paraffin, cut into 5-µm sections and stained with haematoxylin and eosin (H&E). Slides were scanned using Olympus Slideview VS200. Tumour burden (percentage tumour area per lung area) in H&E-stained sections of all five lung lobes was quantified using BioPix iQ software (v.2.1.4). Histological tumour grading was done on non-necrotic lesions according to a previously described protocol in ref. 10. For IHC analyses of mouse and human lung sections as well as tissue microarray (TMA) sections from a Swedish NSCLC cohort37, deparaffinization was followed by epitope retrieval and blocking of endogenous peroxidase activity with H2O2. Sections were then incubated with primary antibodies listed in Supplementary Table 4. Slides were scanned at ×40 on a Hamamatzu Nanozoomer (2.0HT). The total number of positively stained cells in tumours was counted and normalized to either the tumour area or the total number of cells.
Cells
Primary tumour cultures were isolated from lung tumours of KP-Young and KP-Old mice roughly 30 weeks following tumour induction. Primary tumour cultures referred to as KP-Y and KP-O were isolated with the tumour dissociation kit for mouse (no. 130-096-730, MACS Miltenyi Biotec) and the gentleMACS Octo Dissociator (no. 130-093-235, MACS Miltenyi Biotec). A549 cell line, a human lung adenocarcinoma epithelial cell line, was obtained from the American Type Culture Collection (catalogue no. CCL-185). Primary cultures as well as the cells were maintained in DMEM medium (Fisher Scientific) supplemented with 0.1% gentamycin (10 mg ml−1, Fisher Scientific), 2% l-glutamine (200 mM, Fisher Scientific) and 10% fetal bovine serum (Thermo Fisher). All cell lines were routinely tested negative for mycoplasma. For the different assays, cells were seeded in Roswell Park Memorial Institute (RPMI) media (Fisher Scientific) supplemented with 10% fetal bovine serum; 1% l-glutamine and 0.1% gentamicin.
Proliferation and viability assays
For population doublings assays, KP-Y1, KP-Y2 and KP-O1, KP-O2 cells were seeded in triplicate in 6-well plates, counted 3 days later and reseeded at the same initial density, for a total of 12–15 days. For cell viability assays, cells were plated in a white, opaque 96-well plate with clear bottom at a density of 2.5 × 103 cells per well in RPMI. 24 h after seeding, drugs were added at the indicated concentrations. Then 72 h after drug addition, cell viability in the presence of all the compounds was assessed by CellTiter-Glo 2.0 (no. G9242, Promega) with a spectrophotometer (Synergy BioTex HTX).
Clonogenic assay
A total of 5,000 KP-Y1, KP-Y2 and KP-O1, KP-O2 cells were seeded in triplicates in 10 cm plates in RPMI media supplemented with 2 mM, 1 mM, 0.5 mM or 0.25 mM concentrations of l-glutamine for 6 days. For clonogenic assays with ISRIB, KP-Y1 and KP-O1 cells were treated with 2 µM ISRIB 3 times a day for 7 days, and a total of 1,000 KP-Y1 and KP-O1 cells were seeded in triplicates in 10-cm plates in DMEM media supplemented with 2 µM ISRIB or vehicle (DMSO) for another 7 days. Colonies were fixed and stained by incubation in PBS containing 0.05% crystal violet, 1% formaldehyde and 1% methanol for 20 min. The number of colonies was counted with OpenCFU62.
3D cultures and anoikis resistance assay
Here 20 × 103 cells were seeded in an ultra-low attachment plate (no. 10023683, Corning) and in parallel in a normal attachment 96-well plate in 3–6 replicates per cell line. For ISRIB experiments, indicated cells were pretreated 3 times a day for 7 days with either ISRIB (2 µM) or vehicle (DMSO) before seeding into ultra-low-attachment plates. After 48 h of cell seeding, cell viability was assessed by CellTiter-Glo 2.0 (no. G9242, Promega) with spectrophotometer (Synergy BioTex HTX). Cell viability values from ultra-low-attachment conditions were normalized to those from adherent plates that was quantified 16 h after seeding.
Spheroid formation and collagen invasion assays
Here 20 × 103 cells were seeded in an ultra-low attachment plate (no. 10023683, Corning) with 10% Matrigel (no. 11523550, Fisher Scientific). For collagen invasion assays, 10 × 103 cells were seeded in an ultra-low attachment plate (no. 10023683, Corning). The next day, cells were embedded in collagen (no. ECM675, Sigma-Aldrich). Images were acquired using Zeiss Axio Observer.A1 reverse microscope with an AxioCam MRm camera (Carl Zeiss) 24–48 h later.
Caspase activity assay
Briefly, 20 × 103 cells were seeded in an ultra-low attachment plate (no. 10023683, Corning) and in parallel in a normal attachment 96-well plate in 5 replicates per cell line. After 48 h of cell seeding, caspase activity in cells was measured using the commercially available Caspase-Glo 3/7 Assay (no. G8090, Promega). Values from ultra-low-attachment conditions were normalized to those from adherent plates that was quantified 16 h after seeding.
Cell trace proliferation assay
Cells were labelled using the CellTrace Proliferation kit (no. C34554, Thermo Fisher Scientific) according to the manufacturer’s instructions and seeded in low attachment plates to form spheroids. Single-cell suspensions were prepared from spheroids on days 0, 2, 4, 6 and 8, flow cytometry data were collected on Sony ID7000 Spectral Cell Analyzer and analysed using Flow Jo software v.10.8.1.
Lentiviral production and transduction
Lentiviruses were produced by transfecting 293FT packaging cells (no. R70007, Life Technologies) with lentiviral backbone constructs, packaging plasmid psPAX2 (Addgene plasmid no. 12260) and envelope plasmid pMD2.G (Addgene plasmid no. 12259) using the X-tremeGENE 9 DNA Transfection Reagent (no. 6365809001, Sigma-Aldrich). Lentiviral supernatants were collected 2 days after transfection. Target cells were transduced once with lentiviruses supplemented with 8 μg ml−1 polybrene (no. TR-1003-G, Sigma-Aldrich) and selected with puromycin (2 μg ml−1, no. 12122530, Fisher Scientific). Luciferase-GFP-expressing vector pLV[Exp]-EGFP:T2A: Hygro-EF1A>Luc2 was generated by VectorBuilder. The GFP expression was validated using flow cytometry. For CRISPR-mediated gene knockout, the LentiCRISPRv2 (Addgene plasmid no. 52961) vector was digested with BsmBI and ligated with BsmBI-compatible pre-annealed oligonucleotides63,64. For overexpression of ATF4 in primary cultures as well as A549, plasmids TFORF3549 and TFORF3036 were a gift from F. Zhang (Addgene plasmids nos. 145025 and 144512)65. The expression of target proteins in CRISPR-knockout and overexpression experiments were evaluated by western blotting 3–5 days after selection. Sequences are provided in Supplementary Table 3.
shRNA-mediated knockdown
Doxycycline-induced knockdown of Atf4 was achieved by cloning miR-E shRNAs targeting Atf4 into the LT3GEPIR vector as previously described in detail in ref. 66. In brief, LT3GEPIR was digested with XhoI and EcoRI, and purified with a gel extraction kit (no. 28704, Qiagen). Single-stranded ultramers were amplified with forward primer miRE-XhoI and reverse primer mirE-EcoRI. Amplicons were gel-purified, digested with XhoI and EcoRI, cleaned up with a PCR purification kit (no. 28104, Qiagen) and ligated into the cut LT3GEPIR vector with T4 DNA Ligase at a 3:1 insert:vector molar ratio. Vectors were transduced into cells and selected with 2 µg ml−1 puromycin for 2 days. Knockdown of ATF4 was verified by western blot analysis following 72 h of treatment with 1 µg ml−1 doxycycline (no. D9891, Sigma-Aldrich). All experiments performed with shRNA-mediated knockdown were performed following 72 h treatment with 1 µg ml−1 doxycycline. Sequences are provided in Supplementary Table 3.
Western blotting
Proteins were isolated by using the 2× Laemmli Buffer (no. 1610737, BioRad) supplemented with β-mercaptoethanol (no. M3148, Sigma-Aldrich). Samples were subsequently heated at 95 °C for 10 min. Proteins were separated on 4–20% Mini-PROTEAN TGX Stain-Free gel (no. 4568096, BioRad) and then transferred onto a 0.2 µM nitrocellulose membrane (BioRad), incubated with specific primary antibodies listed in Supplementary Table 4. Protein bands were detected using Clarity Western ECL substrate (no. 1705061, BioRad) with the Amersham ImageQuant 800 western blot imaging systems (cytiva). A list of antibodies used is provided in Supplementary Table 4.
Real-time quantitative PCR
RNA was isolated with the RNeasy Plus Mini kit (no. 74136, Qiagen), and complementary DNA (cDNA) was synthesized using iScript Adv cDNA Kit for RT–qPCR (no. 1725038, BioRad). Gene expression was analysed using the PowerUp SYBR Green Master Mix (no. A25777, Thermo Fisher Scientific) on QuantStudio 5 Real-Time PCR system (Thermo Fisher). The list of primers used is provided in Supplementary Table 3.
Reagents
Cells attached or in spheroids were treated with indicated drugs at the following concentrations, unless stated otherwise in the figure: vehicle (DMSO D8418, Sigma-Aldrich), 100 nM CB-839 (5337170001, Sigma-Aldrich), 2 mM pyruvate (11501871; Fisher Scientific); 50 µM Trolox (648471, Sigma-Aldrich); 2 mM DMG (349631, Sigma-Aldrich), 500 µM N-acetyl-l-cysteine (A7250, Sigma-Aldrich), 0.3 µM erastin (E7781, Sigma-Aldrich); 20 µM 5-methyltetrahydrofolic acid disodium salt (M0132, Sigma-Aldrich); 5 µM hypoxanthine (H9636, Sigma-Aldrich), 1× nucleosides (ES-008-D, Sigma-Aldrich), 1× non-essential amino acids (SH30238.01, Nordic Biolabs), 2 mM l-serine (S4311, Sigma-Aldrich), 2 mM l-asparagine (A4159, Sigma-Aldrich), 2 µM folic acid (F8758, Sigma-Aldrich), 50 µM thymidine (T1895, Sigma-Aldrich), 1 mM sodium formate (247596, Sigma-Aldrich), 20 µM l-aspartic acid (A7219, Sigma-Aldrich), 2 µM ISRIB (HY-12495, MedChemExpress), bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl) ethyl sulfide (BPTES, HY-12683, MedChemExpress), V-9302 (HY-112683, MedChemExpress), ERG240 (HY-W193545A, MedChemExpress), branched-chain aminotransferase inhibitor (HY-116044, MedChemExpress), NCT-503 (HY-101966, MedChemExpress), l-6-diazo-5-oxonorleucine (DON, HY-108357, MedChemExpress), 2-DG (Sigma-Aldrich), 10 µM MG-132 (HY-13259; MedChemExpress), 50 ng ml−1 actinomycin D (SBR00013-1ML; Sigma-Aldrich), 20 µg ml−1 cycloheximide solution (C4859, Sigma-Aldrich), 5 µM or 1 µM of PERK inhibitor (GSK2656157, HY 13820, MedChemExpress).
Mitochondrial respiration
Oxygen consumption rate (OCR) and extracellular acidification rate experiments were performed using the XF96pro apparatus from Seahorse Bioscience (Agilent). Cells were seeded to roughly 90% confluence for each condition in RPMI. The following day, media was completely replaced with RPMI pH 7.4 (103576-100, Agilent) containing 10 mM glucose and 2 mM glutamine and incubated for 45 min at 37 °C in a CO2-free incubator before measurements. Basal and maximal respiration measurements were obtained by performing a mito-stress test (103015-100, Agilent) following the manufacturer’s instructions. Data were normalized to protein content using Pierce BCA Protein Assay Kit (23225, ThermoScientific).
GC–MS analysis of polar metabolites and stable isotope tracing
For analysis of tumour primary cultures in attached conditions, 2.5 × 105 cells were seeded in 6-well plates containing 2 ml of stable isotope RPMI media supplemented with 10% dialysed fetal bovine serum with 2 mM [U13C]-l-glutamine (Cambridge Isotope Laboratories), 10 mM [1,2-13C]-d-glucose (Sigma-Aldrich) or [3-13 C]-d-glucose (Sigma-Aldrich) for 8 h. Cells were washed twice in ice-cold saline and then collected by scraping in 200 µl of 80% (v/v) ice-cold methanol containing 1 µg ml−1 norvaline (Sigma-Aldrich). Samples were then vortexed for 10 min at 4 °C and then centrifuged at maximum speed for 10 min. The supernatant was transferred to fresh tubes and then dried in a speed vac. Dried metabolite extracts were then derivatized with 20 ml of O-methoxyamine-hydrochloride reagent (89803, Sigma-Aldrich) in pyridine (270407, Sigma-Aldrich) at a concentration of 20 mg ml−1 for 60 min at 37 °C and 20 ml of N–tert-butyldimethylsilyl-N-methyltrifluoracetamide with 1% tert-butyldimethylchlorosilane (375934, Sigma-Aldrich) for 60 min at 37 °C. After derivatization, samples were analysed by gas chromatography with mass spectrometry (GC–MS) using a DB-35ms column (Agilent Technologies) in an Agilent Intuvo gas chromatograph coupled to an Agilent 5977B mass spectrometer. Helium was used as the carrier gas at a flow rate of 1.2 ml min−1. Then 1 µl of sample was injected in split mode (split 1:1) at 270 °C. After injection, the GC oven was held at 100 °C for 1 min and then increased to 300 °C at 3.5 °C min−1. The oven was then ramped to 320 °C at 20 °C min−1 and held for 5 min at 320 °C. The MS system operated under electron impact ionization at 70 eV and the MS source and quadrupole were held at 230 °C and 150 °C, respectively, the detector was used in scanning mode, and the scanned ion range was 10–650 m/z. Mass isotopomer distributions were determined by integrating the appropriate ion fragments for each metabolite67 using MATLAB (Mathworks) and an algorithm adapted from ref. 68 that corrects for natural abundance. For all data, total or relative metabolite pool sizes are normalized to cell counts for each condition.
RNA-seq
Total RNAs were extracted with the RNeasy Plus Mini kit (74136, Qiagen). RNA quality was assessed using the DNF-471 RNA kit on a fragment analyser (Agilent). RNA-seq libraries were prepared according to the Smart-seq2 protocol, developed69 with some minor modifications. Reverse transcription followed by preamplification of purified cDNA was performed as described in ref. 70. Samples were purified using Agencourt AMPure XP beads (BD Bioscience). Purified samples were further used for library preparation with the Nextera XT DNA library preparation kit and Nextera XT index kit v2 (Illumina), according to the manufacturer’s recommendations. Samples were indexed and amplified by adding 15 µl of Nextera PCR master mix and 5 µl of each index, i7 and i5, obtaining a reaction volume of 50 µl. Amplification and indexing were performed in a T100 instrument at 95 °C for 10 s, 55 °C for 30 s, and as a final step an incubation at 72 °C for 5 min.
Using Agencourt AMPure XP (BD Bioscience) samples were again purified by following the manufacturer’s protocol. Concentrations were measured using Qubit double-stranded DNA high sensitivity Assay Kit, on a Qubit 4 fluorometer (Invitrogen, Thermo Fisher Scientific). The quality control and the fragment size distribution were determined using the DNF-474 High sensitivity next-generation sequencing kit on a Fragment analyser (Agilent).
Libraries were pooled and sequencing was performed at the Genomics core facility at the University of Gothenburg on a NextSeq 500 instrument (Illumina) using the NextSeq 500/500 High Output Kit v.2.5 (150 cycles) and paired-end sequencing (2 × 75 cycles).
Indexing of the ENSEMBL GRCm39 reference genome as well as alignment of sequencing reads was performed using STAR version 2.7.9a (ref. 71). Read count of aligned reads was performed with HTSeq version 0.9.1 (ref. 72). Before differential expression analysis, genes with an average read count of less than 3 were excluded. Differential expression analysis was performed with DESeq2 version 1.34.0 (ref. 73) in R version 4.1.2. First, the four replicate samples within each cell line were merged using the collapseReplicates function. Subsequently, the analysis was run comparing the two groups of primary cultures, Young and Old as well as between KP-Y o/e and KP-Y control. Significantly regulated genes were defined as having at least twofold regulation with an adjusted P value (Benjamini–Hochberg) less than or equal to 0.05. GSEA was done using the GSEA desktop v.4.3.3 (ref. 74). Functional annotation of genes by Gene Ontology and pathway analysis by Kyoto Encyclopedia of Genes and Genomes were done using DAVID75. Protein–protein association data was obtained from the STRING database (v.12.0) and integrated to analyse both functional and physical interactions using curated datasets and high-throughput experimental evidence. Computational predictions and experimental data were combined to construct and score protein networks, which was subsequently analysed for enriched biological functions and potential input biases.
ATAC-seq
Library construction for ATAC-seq of paired-end 150-bp-long reads by the Illumina HiSeq was performed at GENEWIZ Azenta Life Sciences. In brief, sequencing adaptors and low-quality bases were trimmed using Trimmomatic v.0.38. Cleaned reads were next aligned to reference genome mm10 using Bowtie2. Aligned reads were filtered using samtools v.1.9 to keep alignments that have a minimum mapping quality of 30. PCR or optical duplicates were marked using Picard v.2.18.26 and removed. Before peak calling, reads mapping to mitochondria (mt) were called and filtered, and reads mapping to unplaced contigs were removed. MACS2 v.2.1.2 was used for peak calling to identify open chromatin regions. Called peaks were filtered for blacklisted regions to mitigate errors due to mappability. Valid peaks from each group were merged and peaks called in at least 66% of samples are kept for downstream analyses. Reads falling beneath peaks were counted in all samples, and these counts were used for differential peak analyses using the R package Diffbind (v.3.20.0). All sequencing tracks, bigWig files were viewed using the Integrated Genomic Viewer (v.2.16.2). GREAT analysis was done to identify the enriched pathways from ATAC-seq.
Western Sweden patient cohort
All patients in western Sweden (eight hospitals) diagnosed with NSCLC from the years 2016 to 2018 and molecularly assessed were included (n = 997). Age at diagnosis, histology and tumour size at diagnosis from computed tomography scans were obtained from patient charts. Patients were assessed with next-generation sequencing for mutational status on DNA from formalin-fixed paraffin-embedded blocks or cytological smears using the Ion AmpliSeq Colon and Lung Cancer Panel v2 from Thermo Fisher Scientific as a part of the diagnostic workup process at the Department of Clinical Pathology at Sahlgrenska University Hospital, assessing hotspot mutations in EGFR, BRAF, KRAS and NRAS.
To obtain the most recent and accurate untreated primary tumour size, measurements were collected from the radiology report of computed tomography performed before a final diagnosis of NSCLC was established. For patients with two or three tumour dimensions available in their charts, tumour volume was calculated using mathematical formulas76. The formula used for tumours with two dimensions is volume = (width × width × length/2), where width is the lowest measurement and length is the highest measurement. For tumours with three dimensions, the tumour volume was calculated as follows: volume = width × length × height. Tumour volume sizes were converted to the same unit of cm3. Pearson correlation was used to identify the linear correlation between tumour volume and age for patients with KRAS mutated and with KRAS wild type. Approval from the Swedish Ethical Review Authority (Dnr 2019-04771) was obtained before the commencement of the study. No informed consent was required because all data were presented in a de-identified form according to the Swedish Ethical Review Authority.
TMA cohort
TMAs from the Swedish NSCLC cohort included primary NSCLC resections (1995–2005)37. All procedures complied with Swedish legislation and were approved by the Uppsala Ethical Review Board (2006/325). All KRAS-mutant lung adenocarcinomas were selected; ATF4 expression was scored and correlation with survival outcomes analysed.
Statistics and reproducibility
GraphPad Prism (v.9.0 and v.10.0) software was used for statistical analyses. Data are presented as mean ± standard error of the mean (s.e.m.), unless otherwise specified. All statistical tests were two-tailed, and replicates represent biological replicates unless otherwise stated. P values were calculated using two-sided Student’s t-tests for measurements of tumour burden or for comparisons between two groups, unless indicated otherwise in the figure legends. For contingency tables, Fisher’s exact test or the chi-squared test was used, as appropriate. One-way analysis of variance (ANOVA) with Tukey’s post hoc test was used for comparisons among several groups, and two-way ANOVA was used for analyses involving several groups across time. Statistical tests used for each experiment are indicated in the corresponding figure legends. A P value less than 0.05 was considered statistically significant. Western blot experiments were replicated at least three times with reproducible results. When representative images are shown, a minimum of three samples per group from the larger cohort were evaluated. All in vitro assays were performed at least three times with a minimum of n = 3 biological replicates per group. For in vivo experiments, the minimum sample size was three independent mice or tumours, with exact sample sizes per condition reported in the figure legends. Spearman’s r was calculated to analyse the correlation of age with EMT marker expression. Patients were divided into two groups according to age at diagnosis and compared for KRAS gene alteration frequencies. No statistical methods were used to predetermine sample size. Individual data points are shown as one dot per sample. Samples and experimental animals were randomly assigned to experimental groups, and sample collection was performed randomly when possible.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
First Appeared on
Source link

