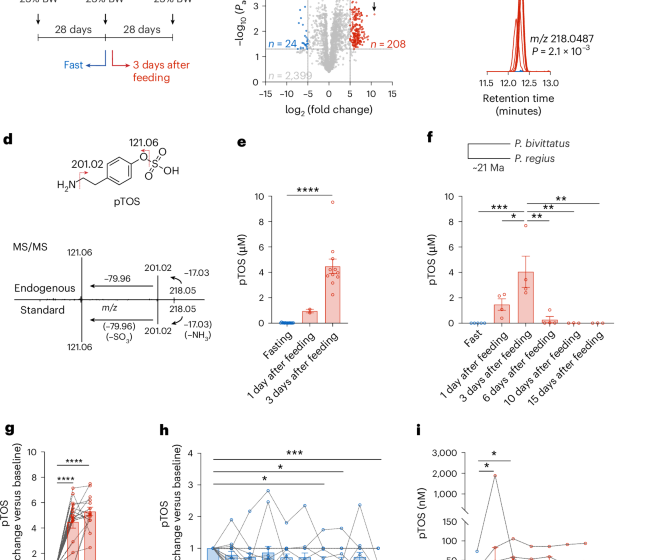
Python metabolomics uncovers a conserved postprandial metabolite and gut–brain feeding pathway
Python husbandry
The animal protocols and procedures involving pythons were approved by the Institutional Care and Use Committee of the University of Colorado Boulder. Captive-bred Burmese pythons (P. molurus bivittatus) and Ball pythons (P. regius) were purchased from Bob Clark Reptiles, Oklahoma City. Pythons were single-housed in the University of Colorado Boulder vivarium in 12-h light–dark cycles at 30 °C with 50% relative humidity. Pythons were then subjected to fasting and feeding schedules as in Fig. 1a, consisting of a 28-day fasting period followed by feeding with a meal equal to 25% of their body weight. Burmese pythons were approximately 2 years old and weighed 1.5–2.5 kg at the start of the study. Ball pythons were approximately 1 year old at the start of the experiments and weighed 400–500 g.
pTOS synthesis and purity
pTOS was synthesized according to the scheme in Supplementary Fig. 1. To a solution of compound 1 (40 g, 291.6 mmol, 1.0 eq) in tetrahydrofuran (THF) (2.0 l) at 0 °C, FmocCl was added (75.43 g, 291.6 mmol, 1.0 eq) in THF (1.0 ml). The mixture was stirred for 16 h. Once compound 1 was consumed completely, the mixture was adjusted to pH 1 with 1 N HCl. The mixture was extracted with ethyl acetate (800 ml ×3). The organic phase was washed with brine and dried over sodium sulfate, filtered and concentrated. The residue was triturated with dichloromethane, filtered and dried to afford compound 2 (49 g, 47%) as white solid. To a solution of compound 2 (10.0 g, 27.8 mmol, 1.0 eq) in dioxane (10 ml) in dimethylformamide (100 ml), SO3-dimethylformamide (12.8 g, 83.4 mmol, 3.0 eq) and pyridine (20 ml) were added. The mixture was stirred for 16 h. Once compound 2 was consumed completely, the residue was adjusted to pH 8 with 5% aqueous NaHCO3 solution. The mixture was concentrated and diluted with water (100 ml). The precipitate was filtered to afford white solid. The solid was purified using C18 (50% acetonitrile in water) and freeze-dried to obtain compound 3 (10.0 g, 78%) as white solid. To a solution of compound 3 (17.0 g, 36.9 mmol, 1.0 eq) in dioxane and water (1:1, 35 mL), Pd(OH)2/C (1.7 g) was added under N2 atmosphere. The mixture was stirred under hydrogen atmosphere for 48 h (15 psi). Once compound 3 was consumed completely, the mixture was filtered through a pad of Celite and the filtration was concentrated. The residue was triturated with ethyl acetate, filtered and dried to afford pTOS (7.7 g, 87%) as white solid with more than 95% purity using 1H NMR (400 MHz) (Supplementary Fig. 2) and more than 96% purity using high-performance LC (Supplementary Fig. 3).
1H NMR (400 MHz, D2O) δ 7.35 (d, J = 8.1 Hz, 2H), 7.29 (d, J = 8.3 Hz, 2H), 3.00 (t, J = 6.9 Hz, 2H), 2.85 (t, J = 7.0 Hz, 2H).
Mouse husbandry
All mouse experiments were performed according to procedures approved by the Institutional Care and Use Committee of Stanford University and Baylor College of Medicine. Mice were maintained in 12-h light–dark cycles at 22 °C with 50% relative humidity. Mice were fed with a standard rodent chow diet (18% protein and 6% fat; Envigo Teklad 2018) or an HFD (60% kcal fat; cat. no. D12492, Research Diets), as specified.
Acute mouse feeding studies
Male mice aged 12–14 weeks (C57BL/6J, strain no. 000664) and 14–15 weeks (C57BL/6J DIO, strain no. 380050) were obtained from The Jackson Laboratory. DIO mice were placed on an HFD for 8–9 weeks since 6 weeks old. Standard rodent chow (18% protein, 6% fat; Envigo Teklad 2018) or an HFD (60% kcal from fat) were provided to lean or DIO mice, as specified. Mice were single-housed 3 h before the onset of the dark cycle with ad libitum access to food and water. At the onset of the dark cycle, mice received a pTOS intraperitoneal injection (50 mg kg−1 or as indicated in the figures) or oral gavage. Food weight was recorded at baseline (0 h) and after 3 h. The body weights of DIO mice were: intraperitoneal vehicle 37.0 ± 2.5 g, pTOS 37.4 ± 0.7 g; oral: vehicle 34.1 ± 1.2 g, pTOS 34.1 ± 1.0 g.
Chronic pTOS treatment in DIO mice
For the chronic treatment, WT C57BL/6J 8-week-old male mice were fed with an HFD (60% kcal from fat) for 8 weeks. The initial body weights of mice were: vehicle 37.8 ± 3.3 g, pTOS 40.0 ± 3.0 g. Mice were then single-housed and administered pTOS (50 mg kg−1, intraperitoneally) or vehicle daily 3 h before the onset of the dark cycle for 28 executive days. The mixture of 18:1:1 (by volume) of saline was Kolliphor EL (cat. no. C5135, Sigma-Aldrich). Dimethyl sulfoxide (DMSO) (Kolliphor) was used as vehicle. Daily food and body weight were recorded.
Python feeding studies
Pythons were randomly assigned to different endpoint groups. They were then fasted for 28 days, after which they received a rat meal equivalent to 20–25% of their body weight. At each pre-assigned endpoint, pythons were euthanized via rapid decapitation under deep isoflurane-induced anaesthesia. Sufficient anaesthetic depth was confirmed by lack of response to physical stimuli. Blood was collected in BD Vacutainer Lithium Heparin tubes (Thermo Fisher Scientific), immediately mixed by inversion, placed on ice for 10 min and then plasma was separated by centrifugation at 3,000g for 15 min to pellet red blood cells. The supernatant was collected, flash-frozen in liquid nitrogen and stored at −80 °C until further analysis. Dissected tissues were rinsed in ice-cold PBS and then flash-frozen in liquid nitrogen and stored at −80 °C until further analysis.
Python oral gavage studies
Ball pythons were randomly assigned to experimental or control groups and then fasted for 28 days. Tyrosine, water or pTOS were then delivered by oral gavage using a 5 Fr. 16-inch rubber Sovereign Sterile Feeding Tube (MWI). Tyrosine was administered at 1 g kg−1 dissolved in water. pTOS was administrated at 50 mg kg−1 in an 18:1:1 mixture of water. Pythons that received pTOS or vehicle control were euthanized and dissected 90 min after gavage. Pythons that received tyrosine were euthanized and dissected 24 h after gavage. Plasma and tissues were collected as described above.
Human meal-test study (Voldstedlund study)
Arterial plasma samples were obtained from a meal-test study that was approved by the Research Ethics Committee of Copenhagen and published previously24. Briefly, ten young male individuals (age: 26.7 ± 1.3 years; BMI: 22.6 ± 0.6 kg m−2) first performed a 60-min one-legged dynamic knee extensor exercise 90 min after a small breakfast (18 kJ per kg) in the morning. Four hours later (time ‘0 min’), individuals received a mixed solid meal (30 kJ per kg body weight), followed by two mixed liquid meals (20 kJ per kg body weight; Nutridrink, Nutrica) 30 min and 60 min after the solid meal. The mixed meals were composed of (energy intake) 50% carbohydrates, 35% fat and 15% protein.
Tyramine production from fecal samples
Fecal samples from ball pythons (21 mg) were dispensed in 500 μl SAAC medium, described previously44. The mixture was centrifuged at 100g for 5 min to pellet undigested materials. Then, 100 μl of the supernatant was mixed with 300 μl SAAC medium and incubated in an anaerobic chamber (Coy Laboratories) at 37 °C for 2 days, in an atmosphere of 5% hydrogen, 10% carbon dioxide and 85% nitrogen. Control SAAC medium contained 1 mM tyrosine and 0.1 mg l−1 vitamin B6. Testing medium contained 50 mM tyrosine and 5 mg l−1 vitamin B6. Cells were pelleted by centrifugation at 3,381g to separate bacteria from the conditioned medium. Then, 150 μl of 2:1:1 acetonitrile:methanol:water mixture was added to extract metabolites from the bacteria and 50 μl of medium were mixed with 150 μl 2:1 acetonitrile:methanol to extract the metabolites from medium.
Tyramine production from the python microbiome
Intestinal content was collected from the small and large intestines of a fed (3 days after feeding) and a fasted (107 days after feeding) python and stored at −80 °C. The intestinal content was then washed in reduced PBS supplemented with 0.05% cysteine and centrifuged at 100g for 5 min to pellet undigested materials. The supernatant was then centrifuged at 5,000g for 5 min to pellet bacteria. Bacteria were resuspended in 10 ml of reduced Gifu medium. A final concentration of 10 g l−1 tyrosine and 500 mg l−1 vitamin B6 was used in the experimental group. Bacteria were cultured for 48 h at 30 °C in an anaerobic chamber. At the end of the incubation period, cells were pelleted by centrifugation at 5,000g for 5 min. The medium was collected for further LC–MS analysis.
Python ABX treatment
A cocktail of ABX containing 200 mg kg−1 metronidazole, 200 mg kg−1 ampicillin, 100 mg kg−1 neomycin and 100 mg kg−1 erythromycin was delivered to juvenile Burmese pythons (weighing 125–200 g at the time of the study) via oral gavage over a 7-day period. The ABX cocktail was administrated to pythons via oral gavage on days 1, 2, 3, 5 and 7. Control pythons received oral gavage of vehicle (water) at an equivalent volume. Concurrently, ABX-treated pythons also received metronidazole, ampicillin, neomycin and erythromycin (each at 0.1 g l−1) in the drinking water. Seven days after the first dose of ABX, pythons were either euthanized or fed a mouse meal equal to 25% of their body weight. ABX-treated pythons received a germ-free mouse meal, whereas vehicle-treated pythons received a specific pathogen-free mouse meal. ABX-treated pythons continued to receive the antimicrobial cocktail via drinking water after their meal. Pythons were euthanized with or without ABX treatment at 3 and 28 days after feeding. Plasma was collected as described above.
Large intestinal content DNA quantification and gel visualization
The large intestinal contents of fasted and 3 days after feeding pythons were emptied; from them, DNA was isolated using the ZymoBIOMICS DNA Miniprep Kit. DNA from 5 mg of intestinal contents was mixed with 3 µl of Novex Hi-Density TBE Sample Buffer and electrophoresed using a 10% (w/v) Novex TBE Gel for 1.5 h at 145 V. The gel was stained using 0.8 µg ml−1 ethidium bromide in water for 5 min; the gel was imaged using the UV setting on a Cytiva IQ 800 gel imager. Lanes within the gel were quantified using Fiji (Image J) and normalized to the average intensity of fasted, vehicle-treated pythons.
TMT-based proteomics of livers from ball pythons
Livers from fasted (n = 5), 1 day after feeding (n = 5) and 3 days after feeding (n = 4) ball pythons were collected as outlined above. Liver samples were homogenized with metal beads in lysis buffer (200 mM EPPS, pH 8.5, 8 M urea, 0.1% SDS, 1× protease inhibitors and 1× phosphatase inhibitors). The supernatant was collected after centrifugation at 21,130g for 10 min. Then, 25 μg of protein from each sample was reduced with tris(2-carboxyethyl)phosphine, alkylated with iodoacetamide and then further reduced with dithiothreitol. Proteins were precipitated onto SP3 beads and digested with Lys-C (1:25) overnight at room temperature, followed by trypsin (1:25) for 6 h at 37 °C. Peptides were labelled with TMT 18plex reagents. The, 2 μl of each sample was pooled and used to shoot a ratio check to confirm complete TMT labelling and to allow for normalization of each sample. All 14 TMTPro-labelled samples were pooled according to the ratios determined from the ratio check. Peptides were desalted using a Sep-pak and fractionated into 24 fractions using basic reverse-phase high-performance LC. Twelve fractions were solubilized, desalted by stage tip and analysed on an Orbitrap Eclipse mass spectrometer with a field asymmetric waveform ion mobility spectrometry device enabled. MS/MS spectra were searched using the COMET algorithm against a Python UniProt composite database containing its reversed complement and known contaminants. Peptide spectral matches were filtered to a 1% false discovery rate using the target-decoy strategy combined with linear discriminant analysis. The proteins were filtered to a less than 1% false discovery rate and quantified only from peptides with a summed signal-to-noise threshold greater than 140. A total of 6,389 proteins were quantified and used in the further analysis.
pTOS production with python liver slices
Fed (3 days after feeding) and fasted (31 days after feeding) ball pythons were euthanized via rapid decapitation while under anaesthesia and the liver was immediately collected and washed in sterile PBS. Freshly isolated liver was transferred to a sterile fume hood and 0.5 g of total liver mass was diced into fine segments (~2 × 2 mm) and then placed into one well of a 6-well plate containing 2 ml William’s E medium supplemented with 1% non-essential amino acids, 1% GlutaMAX, 2% fasted python plasma, 100 nM dexamethasone, 100 nM insulin and 0.375% fatty-acid-free bovine serum albumin. Tyramine (1 mM final concentration) or vehicle control was added to the medium and incubated at 30 °C, 5% CO2 for 16 h. The medium was collected and clarified by centrifugation at 21,130g for 10 min. The supernatant was flash-frozen in liquid nitrogen and stored at −80 °C until further analysis.
Python SULT expression in HEK 293T cells and enzymatic assay
Aryl SULTs with higher abundance in postprandial python livers were codon-optimized for expression in HEK 293T cells and synthesized by Integrated DNA Technologies. The sequences were then cloned into a pCMV5-mCherry vector with a 3× FLAG tag on the C terminal. For simplicity, we named the python SULTs with the following annotations based on the sequence similarities: SULT1A1 (A0A9F5IVD0), SULT1C4 (A0A9F2QWK4), SULT1D1 (A0A9F2REM3) and SULT6B1 (A0A9F2R4U1). HEK 293T cells were transiently transfected with Polyfect; 24 h later, cells were washed with ice-cold PBS and scraped into an Eppendorf tube. Cells were pelleted using centrifugation at 845g for 5 min at 4 °C and lysed in 50 mM potassium phosphate buffer (pH 7.4) using sonication. The protein concentration of the soluble fraction was determined using a BCA assay. Tyramine SULT activity was determined with cell lysates containing 100 μg total protein, supplemented with 100 μM tyramine, 100 μM PAPS (cat. no. ES019, R&D Sytems) and 1 mM pargyline (cat. no. 10007852, Cayman), a monoamine oxidase inhibitor. The assay was initiated by incubation at 30 °C for 30 min and stopped by adding ice-cold acetonitrile:methanol. SULT expression was validated using immunoblotting. SULT1D1 was not expressed in HEK 293T cells because of its incomplete sequence in UniProt. The activity was also indistinguishable from non-transfected controls. Heat-inactivated cell lysates exhibited non-detectable enzymatic activity.
Tyramine and pTOS extraction from cultured medium for LC–MS
To extract metabolites from the medium of cultured hepatocytes or bacteria, 200 µl of 1-butanol was added to 500 µl of medium. The mixture was vortexed for 1 min and then centrifuged at 4 °C for 10 min at 21,130g. The top layer was carefully transferred to mass spec vials for LC–MS analysis.
Mouse blood and plasma sample preparation for LC–MS
Blood from mice was collected by submandibular bleeding into lithium heparin tubes (cat. no. 365985, Becton Dickson) and immediately kept on ice. Blood was then centrifuged at 4 °C for 5 min at 2,348g. Plasma was transferred into new Eppendorf tubes and stored at −80 °C if not used immediately. Metabolites were then extracted by adding 150 µl of a 2:1 mixture of acetonitrile:methanol to 50 µl of serum or plasma.
CSF collection and preparation for LC–MS
Mice were anaesthetized using isoflurane (cat. no. R510-22, RWD Life Science), with a 5% concentration for induction and 2% for maintenance. Once properly anaesthetized, the mouse’s head was secured onto a custom-made surgical frame in a vertical downward position to locate the cisterna magna. A sagittal incision was made along the midline of the skin, and the subcutaneous tissue and neck muscles were carefully separated to expose the cisterna magna. A 34 G needle (cat. no. 207434-10, Hamilton), connected to a Hamilton syringe (cat. no. 7643-01), was carefully inserted into the cisterna magna after piercing the atlanto-occipital membrane. CSF was slowly withdrawn from each mouse. Then, 30 μl 2:1 acetonitrile:methanol was added to 10 μl CSF to extract the metabolites in CSF.
Tyramine and pTOS measurements in the mouse brain
WT C57BL6/J 11–12-week-old male mice were injected with pTOS (50 mg kg−1) or tyramine (28.7 mg kg−1) or vehicle at the same molarity. Mice were anaesthetized 30 min after the administration and the brain was perfused with ice-cold PBS via cardiac perfusion. The brain was carefully dissected out and water was added at 2:1 volume-to-weight ratio (that is, 800 μl water added to 400-mg brain). After homogenization, 50 μl of the supernatant was mixed with 150 μl 2:1 acetonitrile:methanol to extract metabolites for LC–MS.
LC–MS analysis
Untargeted metabolomics measurements were performed using an Agilent 6545 Quadrupole time-of-flight LC–MS instrument. MS analysis was performed using electrospray ionization in both positive and negative modes. The dual electrospray ionization source parameters were set as follows: the gas temperature was set at 250 °C with a drying gas flow of 12 l min−1 and the nebulizer pressure at 20 psi; the capillary voltage was set to 3,500 V; and the fragmentor voltage was set to 100 V. Separation of metabolites was conducted using a Luna 5 μm NH2 100 Å LC column (cat. no. 00B-4378-E0, Phenomenex) with normal phase chromatography. Mobile phases in positive mode were: buffer A, water with 0.1% formic acid; buffer B, acetonitrile with 0.1% formic acid. Mobile phases in negative mode were: buffer A, 95:5 water:acetonitrile with 0.1% ammonium hydroxide and 10 mM ammonium acetate; buffer B, acetonitrile. The LC gradient started at 100% buffer B with a flow rate of 0.7 ml min−1 from 0 to 2 min. The gradient was then linearly increased to 50% buffer A and 50% buffer B at a flow rate of 0.7 ml min−1 from 2 to 20 min. From 20 to 25 min, the gradient was maintained at 50% buffer A and 50% buffer B at a flow rate of 0.7 ml min−1.
Quantification of metabolite concentration was performed by generating a standard curve with known concentrations of each metabolite. Metabolite standards were analysed alongside the samples using the same method. A standard curve generated from the metabolite concentrations and the extracted ion intensities was used to calculate the concentrations of each metabolite.
TSE PhenoMaster metabolic studies in mice
Sixteen-week-old C57BL6/J male mice were acclimated into the TSE PhenoMaster Metabolic Cage system. In the TSE PhenoMaster cages, mice were maintained on a chow diet for the first 3 days and then fasted for 24 h followed by injection with vehicle or pTOS (50 mg kg−1, intraperitoneally) and refeeding at 5:30 before the start of the night cycle, while food intake, RER, energy expenditure and locomotion were continuously monitored. Energy expenditure data were analysed with each animal’s body weight as a covariate using the online CalR tool45. A mixture of 18:1:1 (by volume) of saline:Kolliphor EL:DMSO was used as vehicle. pTOS was dissolved in the mixture, then aliquoted and stored at −80 °C before use.
Conditioned flavour avoidance
Eight-week-old mice were habituated to restricted water access for 2 h per day (6:30 to 8:30) for 7 days. Food was available ad libitum. Training consisted of two training days. On each training day, mice had access to two burettes containing the same flavour of Kool-Aid (cherry or grape) during the fluid access period of 2 h. Immediately after, each mouse received an intraperitoneal injection of saline, pTOS (50 mg kg−1) or LiCl (95 mg kg−1). LiCl (cat. no. L9650, Sigma-Aldrich) was dissolved in saline and used as the positive control. The order of drug/saline exposure and paired flavours was counterbalanced on different days. Each training day was followed by a non-injection day when water was available during the 2-h fluid access period. Two days after the final training day, animals were provided with both flavours (one flavour per burette) during the 90-min fluid access period. Fluid intake was recorded for 2 h. Th pTOS or LiCl preference was calculated by dividing the consumption of pTOS or LiCl-paired flavour by the total consumption of cherry-flavoured and grape-flavoured water.
Sucrose preference test
Twelve-week-old C57BL6/J male mice were habituated with two bottles of water for 2 days. Mice were then water-deprived for 24 h and then provided with a free choice of either drinking 1% sucrose solution or double-distilled water for 2 h (6:30 to 8:30). Sucrose preference was calculated by dividing the consumption of sucrose by the total consumption of water and sucrose.
Hormone measurements
Twelve-to-fourteen-week-old DIO male mice were administered pTOS (50 mg kg, intraperitoneally) or vehicle control in the light phase (Zeitgeber time 6), with ad libitum access to food and water during the experiment. Blood was collected from mice at the 1-h and 3-h time points. The following hormones were measured with commercially available enzyme-linked immunosorbent assay kits according to the manufacturers’ instructions: ghrelin (cat. no. EZRGRT-91K, Merck Millipore), leptin (cat. no. 90030, Crystal Chem), adiponectin (cat. no. 80569, Crystal Chem), insulin (cat. no. 62100, Crystal Chem), GLP-1 (cat. no. 81508, Crystal Chem) and GDF15 (cat. no. MGD150, R&D Systems).
Measurement of blood glucose levels after pTOS injection
Male C57BL/6 mice aged 8–10 weeks were fasted for 6 h from 10:00 to 16:00 and then given intraperitoneal injections of pTOS (50 mg kg−1, n = 7) or vehicle (n = 7). Blood glucose levels were measured at 0, 20, 40, 60, 90 and 120 min after the injections with a glucometer.
Blood pressure measurements
Mice were anaesthetized with isoflurane and placed in the supine position with the fur removed in the chest area. Blood pressure was measured with a 1.4-F pressure sensor mounted Millar catheter (SPR-671, ADInstruments) inserted into the right carotid artery. Blood pressure was recorded with LabChart 7 Pro (ADInstruments) and annotated with the BP_annotate package in MATLAB45,46.
Oral glucose tolerance test with pTOS injection
Male C57BL6/J mice aged 15 weeks were fasted for 4 h from 10:00 to 14:00. Mice received intraperitoneal injections of pTOS (50 mg kg−1, n = 5) or vehicle (n = 5) at 13:30 and then received glucose by oral gavage (1.5 g kg−1) at 14:00. Blood glucose levels were measured at 0, 20, 40, 60, 90 and 120 min after glucose oral gavage with a glucometer.
Oral lipid tolerance test with pTOS injection
Male 13-week-old C57BL6/J mice were fasted for 4 h from 9:30 am to 13:30. Mice received intraperitoneal injections of pTOS (50 mg kg−1, n = 5) or vehicle (n = 5) at 13:00, and then received corn oil by oral gavage (15 µl g−1) at 13:30. Blood plasma was collected at 0, 1, 2, 4, 6 and 8 h after oral gavage and triglyceride levels were measured with a colorimetric assay kit (cat. no. 10010303, Cayman).
Oral protein tolerance test with pTOS injection
Male C57BL6/J mice aged 15 weeks were fasted for 4 h from 9:30 to 13:30. Mice received intraperitoneal injections of pTOS (50 mg kg−1, n = 5) or vehicle (n = 5) at 13:00, and then received ISOPURE protein powder by oral gavage (1.5 kcal kg−1) at 13:30. Blood plasma was collected at 0, 0.5, 1, 2 and 4 h after oral gavage. Branched chain amino acids (that is, leucine, isoleucine and valine) were used as markers of protein intake and measured with LC–MS.
Gastric emptying assay
Male C57BL/6 mice aged 12 weeks were fasted for 12 h and then given intraperitoneal injections of pTOS (50 mg kg−1, n = 5), exenatide (100 μg kg−1, n = 4) or vehicle (n = 5). After a 30-min interval, animals received 300 μl of a phenol-red-based test meal (50 mg phenol red dissolved in 100 ml of 1.5% carboxymethylcellulose, maintained at 37 °C with gentle stirring) via oral gavage. Mice were euthanized either immediately after gavage (t = 0) or 30 min after gavage (t = 30). Stomachs were excised, homogenized in 25 ml of 0.1 N NaOH and allowed to stand for 1 h at room temperature. From the resulting mixture, 8 ml of supernatant was combined with 1 ml of 33% trichloroacetic acid, followed by centrifugation at 845g for 30 min at 4 °C. The clarified supernatant was then neutralized with 2 ml of 2 N NaOH and the phenol red content was quantified by measuring absorbance at 560 nm. Gastric emptying was calculated according to the following equation: gastric emptying = 100×(1-X/Y), where X represents the mean absorbance from animals euthanized at t = 30 and Y represents the mean absorbance from animals euthanized at t = 0.
TRAP2 mice
We crossed TRAP2 mice (cat. no. 030323, The Jackson Laboratory) with Rosa26-LSL-tdTomato mice (cat. no. 007905, The Jackson Laboratory) to generate TRAP2/Rosa26-LSL-tdTomato or TRAP2 mice. Mice were housed in a temperature-controlled environment using a 12-h light and 12-h dark cycle. Mice were individually housed at least 1 week before the study. Mice were fed a standard chow diet (19.0% protein, 6.5% fat, 2.7% crude fibre, 12.3% neutral detergent fibre, by weight, Harlan Teklad, cat. no. 2920). Water was provided ad libitum.
TRAP induction
We dissolved 4-OHT (cat. no. H6278, Sigma-Aldrich) at 20 mg ml−1 in ethanol by sonication at 37 °C for 15 min. The dissolved 4-OHT was then stored in aliquots at −80 °C for up to several weeks or used immediately. Before use, 4-OHT was dissolved by shaking at 37 °C for 10 min, then sunflower seed oil and castor oil (4:1) was added for a final concentration of 10 mg ml−1. After evaporating the ethanol in a vacuum (845g, 15 min), the final 4-OHT solution was injected intraperitoneally at 50 mg kg−1. To TRAP pTOS-activated neurons, TRAP2/Rosa26-LSL-tdTomato 10-week-old male mice were fasted from 12:00 to 17:00, then received intraperitoneal injection of pTOS (50 mg kg−1), followed by 4-OHT injection (50 mg kg−1, intraperitoneally) 30 min after; 2 weeks later, mice were perfused with saline followed by 10% formalin. As controls, another group of TRAP2/Rosa26-LSL-tdTomato male mice received intraperitoneal injection of vehicle, followed by 4-OHT injection (50 mg kg−1, intraperitoneally) 30 min after; 2 weeks later, mice were perfused. A mixture of 18:1:1 (by volume) of saline:Kolliphor EL:DMSO was used as vehicle. Coronal brain sections were cut at 30 μm and collected into five consecutive series. Sections were cover-slipped and analysed using a fluorescence microscope. The numbers of tdTomato-labelled (TRAPed) neurons were counted and quantified manually. Briefly, for each mouse brain structure analysed, anatomically defined reactive oxygen species (ROIs) were selected based on the Allen Mouse Brain Atlas. The same ROI boundaries were consistently applied across all mice and sections. To ensure consistency, a single coronal brain section per region was analysed for each mouse, selected at matched anterior-posterior coordinates based on anatomical landmarks. Three mice were included in each group.
Chemogenetic approaches
Eight-week-old Male TRAP2 mice were anaesthetized (with 2% isoflurane) and placed in a stereotaxic instrument. Artificial eye ointment was applied to prevent corneal drying, and a heat pad was used to hold the body temperature at 37 °C. To chemogenetically inhibit pTOS-activated PVH or VMH neurons, we injected AAV8-DIO-hM4Di-mCherry (titre: 5 × 1012 GC per ml, 0.2 µl, cat. no. 44362, Addgene) into the PVH or VMH of TRAP2 mice (PVH: −0.82 mm; mediolateral: 0.25 mm; dorsoventral: −4.75 mm; VMH: −1.7 mm; mediolateral: 0.3 mm; dorsoventral: −5.6 mm), respectively. After allowing 2 weeks for virus expression, mice were housed singly before they were subjected to any studies. Mice were fasted overnight (from 17:00 to 9:00), then received saline or CNO (intraperitoneally, 3 mg kg−1, cat. no. 16882, Cayman) injections. After saline or CNO injection, pre-weighed regular chow (6.5% fat; cat. no. 2020, Harlan Teklad) was put back into the cages. Food intake was monitored for 3 h. At the end of the experiments, all mice were perfused with saline followed by 10% formalin. Brain sections were collected and sectioned at 30 µm. The expression of mCherry was examined with histology; only those with accurate targeting were included in the data analyses.
cFos mapping of pTOS-activated neurons in python brains
To process python brains, we adapted the approach as previously described for in vivo fixation of the lizard brain47. One hour after delivery of 50 mg kg−1 pTOS via oral gavage, ball pythons were anaesthetized via isoflurane inhalation for 30 min. Sufficient anaesthetic depth was determined by lack of response to a physical stimulus. The thoracic cavity and rostral ~5 cm were then opened with surgical scissors to expose the heart and carotid arteries. Fine incisions were made in the ventricle and right atrium, and a perfusion needle was inserted into the ventricular incision, through the right aorta and into the left carotid artery. The needle was clamped in place with a haemostat and 50 ml of heparinized PBS (10 units per litre) was perfused using a peristaltic pump to clear blood from the brain. Brain fixation was performed by perfusion with 50 ml of 4% paraformaldehyde (PFA). The brain was then extracted using a corneoscleral punch and submerged overnight in 4% PFA at 4 °C. The brain was removed from PFA, carefully rinsed twice with PBS and then added to a 30% sucrose PBS solution and stored at 4 °C for 2 days to dehydrate the tissue. Coronal brain sections were cut at 30 µm and collected into five consecutive series. One series of the sections was blocked for 1 h in 0.3% PBS with Tween 20 with 5% normal donkey serum. To detect cFos expression, a Rabbit anti-cFos antibody (1:500 dilution, cat. no. 226008, Synaptic System) was added and incubated at 4 °C overnight on shaker. The cFos antibody recognizes the amino acid sequence ‘MFSGFNADYEASSSR’. Three of the 15 amino acids in this sequence differ from those in the corresponding sequence in the python sequence. The following day, slices were rinsed with 0.1% PBS with Tween 20 for 6× 10 min and then incubated with donkey anti-rabbit Alexa Fluor 488 (1:500 dilution, cat. no. A21206, Invitrogen) at room temperature for 2 h. Sections were cover-slipped and analysed using a fluorescence microscope. The numbers of cFos-labelled neurons were counted and quantified manually. The ROIs for each ball python brain structure were selected using the most reliable anatomical landmarks available from the limited existing references. For each ball python, 3–4 coronal brain sections per VMH were analysed. Four pythons were included in each group. The locations of cFos+ neurons in the VMH and other brain regions were mapped onto the standard anatomical brain sections of P. regius34. Furthermore, brain sections from both the same species of red-sided garter snake48,49 and different species with similar brain structures, such as the tree lizard Urosaurus ornatus50, revealed consistent VMH locations. Within the Squamata order, the VMH is one of the brain regions having the least variation, as determined by calculating the log-transformed coefficient of variation for each brain region to assess individual differences51.
Slice electrophysiology
Electrophysiology recordings were performed as described previously 52. Briefly, mice were deeply anaesthetized with isoflurane and transcardially perfused with a modified ice-cold sucrose-based cutting solution (pH 7.3) containing 10 mM NaCl, 25 mM NaHCO3, 195 mM sucrose, 5 mM glucose, 2.5 mM KCl, 1.25 mM NaH2PO4, 2 mM Na-pyruvate, 0.5 mM CaCl2 and 7 mM MgCl2, bubbled continuously with 95% O2 and 5% CO2. Mice were then decapitated and the entire brain was removed and immediately submerged in the cutting solution. Coronal brain slices (220 μm) containing the PVH or VMH were cut with a Microm HM 650V vibratome (Thermo Fisher Scientific) in oxygenated cutting solution. Slices were then incubated in oxygenated artificial CSF (126 mM NaCl, 2.5 mM KCl, 2.4 mM CaCl2, 1.2 mM NaH2PO4, 1.2 mM MgCl2, 11.1 mM glucose and 21.4 mM NaHCO3, balanced with 95% O2/5% CO2, pH 7.4) to recover ~25 min at 32 °C and subsequently for 1 h at room temperature before recording. Slices were transferred to a recording chamber and allowed to equilibrate for at least 10 min before recording. Slices were superfused at 32 °C in oxygenated artificial CSF at a flow rate of 1.8-2 ml min−1. mCherry or tdTomato-labelled neurons were visualized using epifluorescence and infrared differential interference contrast imaging on an upright microscope (Eclipse FN-1, Nikon) equipped with a movable stage (MP-285, Sutter Instrument). Patch pipettes with resistances of 3–5 MΩ were filled with intracellular solution (pH 7.3) containing 128 mM K-Gluconate, 10 mM KCl, 10 mM HEPES, 0.1 mM EGTA, 2 mM MgCl2, 0.05 mM Na-GTP and 4 mM Mg-ATP. Recordings were made using a MultiClamp 700B amplifier (Axon Instrument), sampled using Digidata 1440A and analysed offline with the pClamp 10.3 software (Axon Instruments). Series resistance was monitored during the recording; values were generally less than 10 MΩ and were not compensated. The liquid junction potential was +12.5 mV and was corrected after the experiment. Data were excluded if the series resistance increased dramatically during the experiment or without overshot for action potential. Currents were amplified, filtered at 1 kHz and digitized at 20 kHz. The current-clamp mode was engaged to measure the neural firing rate and resting membrane potential at the baseline or in response to CNO (10 µM). To measure the effects of pTOS on VMH and PVH neurons, the current-clamp mode was engaged to test the neural firing rate and resting membrane potential at the baseline and after bath application of pTOS (1 µM concentration as indicated in the figures). To test if pTOS directly activated VMH neurons, VMH neurons were pretreated with a cocktail of synaptic blockers containing 30 μM CNQX, 30 μM D-AP5 and 50 μM bicuculline to block the excitatory and inhibitory synaptic inputs in the recorded VMH neurons; pTOS-induced responses were recorded as described above.
Statistics
The minimum sample size was predetermined by the nature of the experiments. For the biochemical measurements, at least 3–4 different mice or pythons per group were used. For the behavioural measurements, 7–9 different mice per group were included. For the histology studies, the same experiment was repeated in at least three different mice or pythons. For the electrophysiological studies, at least six different neurons from three different mice were included. The data are presented as the mean ± s.e.m. or as individual data points. Statistical analyses were performed using Prism (GraphPad Software) to evaluate the normal distribution and variations within and among groups. The methods of the statistical analyses were chosen based on the design of each experiment and are indicated in the figure legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
First Appeared on
Source link

