Structural energetics of cold sensitivity
Cloning, cell culture and protein expression
N-terminally fused mEGFP constructs of TRPM8 were obtained using Gibson assembly to sub-clone TRPM8 genes into a pFastBac1 vector. Mutagenesis was carried out by either PCR with mutagenic primers, or by Gibson assembly, with Q5 high-fidelity polymerase. Construct sequences were verified using Sanger sequencing and whole-plasmid sequencing. Expi293F cells were maintained at 37 °C with 8% ambient CO2 and grown to a density of 3–3.5 × 106 cells ml−1. To transduce cells for protein overexpression, a baculovirus (generated from two rounds of viral amplification in SF9 cells, according to known protocols) for the N-terminally fused mEGFP Pm or Hs TRPM8 with a human rhinovirus 3C proteolysis site was added dropwise, for a final concentration of 5–10% (vol%/vol%) to the flask while gently mixing the culture. Baculoviruses used in mammalian cell cultures were sequenced routinely by amplifying the coding region using PCR (M13 primer sites), gel extracting the amplicon and subjecting the purified DNA fragments to Sanger sequencing. Virally transduced Expi293F cultures were left at 37 °C with 8% CO2 for 16 h. To boost expression, the cultures were supplemented with 10 mM sodium butyrate and transferred to an incubator at 30 °C with 5% CO2 and allowed to incubate for a total of 72 h (post-transduction). Cells were then collected by centrifugation (3,000g, 10 min, 4 °C), washed once with 1× Dulbecco’s phosphate buffered saline (DPBS) pH 7.40 by gently resuspending the cell pellet, and collected by centrifugation (3,000g, 10 min, 4 °C). Cell pellets were flash frozen in liquid nitrogen and kept in a −80 °C freezer until use.
Detergent purification for HDX–MS and cryo-EM samples
All purification steps were carried out at 4 °C or over ice, unless otherwise noted. The avian or human TRPM8 channel was purified with the same general protocol. Briefly, a cell pellet grown from 0.5 l Expi293F cell culture (typically 7–10 g in total) was resuspended into lysis buffer (50 mM HEPES-NaOH pH 7.40, 150 mM NaCl, 50 μg ml−1 DNase I, 10 μg ml−1 RNase, 0.2 mM AEBSF, 50 μg ml−1 soy trypsin inhibitor, 10 μg ml−1 leupeptin, 10 μg ml−1 pepstatin, 1 mM benzamidine-HCl and aprotinin) for a total dilution of 4:1 lysis buffer:cell pellet. The resuspension was adjusted to 0.5% lauryl maltose neopentyl glycol (LMNG)/0.5% glycodiosgenin (GDN), and rotated gently for 1 h at 4 °C. After centrifugation (35,000g, 30 min, 4 °C), the lysate was applied to 1 ml anti-GFP-nanobody-conjugated Sepharose 4B resin (prepared in-house) for 2 h at 4 °C with gentle rotation. The TRPM8-bound resin was washed extensively with wash buffer (20 mM HEPES-NaOH pH 7.40, 150 mM NaCl, 0.05% GDN), then digested in the presence of 10 μg ml−1 PreScission protease (prepared in-house) along with 1 mM dithiothreitol for 2 h. The eluate was concentrated to 0.2 ml and injected onto a Superose 6 Increase 10/300 GL column pre-equilibrated in SEC buffer (20 mM HEPES-NaOH pH 7.40, 150 mM NaCl, 0.005% GDN). For the Pm structures obtained at pH 9, cells were resuspended into lysis buffer containing 50 mM Tris-HCl pH 9.0, 150 mM NaCl, 5 mM CaCl2, 50 μg ml−1 DNase I, 10 μg ml−1 RNase, 0.2 mM AEBSF, 50 μg ml−1 soy trypsin inhibitor, 10 μg ml−1 leupeptin, 10 μg ml−1 pepstatin, 1 mM benzamidine-HCl and aprotinin. The resuspended cells were extracted using 0.5% LMNG/0.5% GDN and further purified with anti-GFP nanobody resin. For cryo-EM, the final sample buffer contained 20 mM TRIS pH 9.00, 150 mM NaCl, 5 mM CaCl2 and 0.0025% GDN.
Cell vesicle purification for cryo-EM
A cell pellet grown from 1.0 l Expi293F cell culture (typically 20–25 g in total) was resuspended into lysis buffer (50 mM HEPES-NaOH pH 7.40, 300 mM KCl) supplemented with 2 mM CaCl2, 50 μg ml−1 DNase I, 10 μg ml−1 RNase, 0.2 mM AEBSF, 50 μg ml−1 soy trypsin inhibitor, 10 μg ml−1 leupeptin, 10 μg ml−1 pepstatin, 1 mM benzamidine-HCl, and aprotinin. The mixture was homogenized roughly using a Dounce homogenizer over ice, and the resuspended cells were adjusted to a final volume of 200 ml. The cells were transferred to a metal beaker over ice and lysed using a probe-tip sonicator (2 min total, 5 s on/15 s off, 60% amplitude). The lysate was clarified by centrifugation (12,000g, 15 min, 4 °C) and the supernatant was filtered immediately through a 0.8-μm mixed cellulose acetate filter. The filtrate was then passed through a 10-ml bed of ion exchange resin (Q-Sepharose FastFlow, Cytiva) packed in a gravity column and pre-equilibrated in lysis buffer. The flowthrough was collected and adjusted to 0.5 mM fluorinated fos-choline 8 (Anatrace), which is below its critical micelle concentration of around 3 mM, for a final volume of 240 ml. Vesicles were batch bound to 5 ml anti-GFP nanobody Sepharose 4B resin (prepared in-house) for 2 h at 4 °C with constant rotation. After binding, the resin was collected by centrifugation (500g, 1 min) and transferred to a gravity column. The affinity resin was washed extensively, and the resuspended beads were adjusted to 10 μg ml−1 PreScission protease (prepared in-house) along with 1 mM dithiothreitol. Bound vesicles were eluted by proteolysis for 2 h, and the eluate was collected and concentrated using an Amicon ultra-centrifugal filter (100 kDa, regenerated cellulose) to a final volume of 0.5 ml. After brief centrifugation (10,000g, 2 min, 4 °C), the sample was injected onto a Superose 6 Increase 10/300 column (Cytiva) pre-equilibrated with the same elution buffer. The void volume peak (typically 8.5–9 ml) was pooled and concentrated to 30 μl using a 0.5-ml Amicon ultra-centrifugal filter (100 kDa, regenerated cellulose) and kept on ice. Concentrated vesicle samples were used immediately for or warmed to 37 °C for 5–10 min before cryo-EM sample preparation.
Cryo-EM grid preparation, screening and data collection
Cryo-EM grids for vesicle samples were prepared using either Quantifoil R1.2/1.3 400 mesh holey carbon grids, Quantifoil R1.2/1.3 300 mesh holy carbon grids coated with ultrathin continuous carbon. Detergent samples were prepared using Quantifoil Au R1.2/1.3 300 mesh holey carbon grids. Grids were glow discharged (15 mA, 30 s) and placed in a Vitrobot Mark IV (FEI Company) set at 4 °C with 100% humidity. For vesicle samples, 2.5 μl concentrated sample was applied directly to grids and, after 5 s of incubation, grids were blotted for 2–5 s and plunge-frozen immediately in liquid ethane cooled in a Dewar of liquid nitrogen. To obtain menthol-bound datasets, vesicles were kept at 4 °C for Pm TRPM8 or ambient temperature (21–24 °C) for the human channel, and a final concentration of 1 mM menthol was added from a stock of 200 mM menthol dissolved in 100% ethanol. After mixing the menthol-treated samples gently at respective temperatures, samples were applied to grids, for a total agonist treatment time of 2–15 min. For detergent samples, 2.5 μl of purified TRPM8 concentrated to 14–16 mg ml−1 was applied directly to grids and, after 10 s of incubation, grids were blotted for 5 s before vitrifying. Grids were transferred and stored under liquid nitrogen until screening or data collection. Grids were screened with a Talos Arctica or Glacios 200 kV cryo-TEM (ThermoFisher Scientific) equipped with a K3 direct detector camera (GATAN), and screening datasets were obtained using SerialEM. Data were collected at the University of California San Francisco (UCSF) Cryo-EM Facility with a Titan Krios cryo-TEM equipped with a K3 camera and Bio Quantum post-column energy filter (counting mode pixel size of 0.8189 Å per pixel after 2× Fourier binning) or at the Janelia Cryo-EM Facility on a Krios equipped with a cold field-emission gun, Selectris X energy filter and Falcon 4i camera (physical pixel size of 0.94). For data collection at UCSF, the zero-loss energy selection slit was set to 10 eV. For vesicle datasets, the target defocus was set at −1.0 to −2.5 µm and, for detergent datasets, the target defocus was set at −0.5 to −2.0 µm.
Cryo-EM data processing and refinement
Dose-weighted, motion-corrected micrographs were obtained using MotionCor2 (ref. 52) and Fourier cropped by a factor of two, or by pre-processing using cryoSPARC53. An initial model of Pm TRPM8 in a membrane was generated in cryoSPARC with a screening dataset obtained on a Glacios cryo-TEM (2,420 micrographs) and refined to approximately 5 Å (440 pixel box size, 0.73 Å per pixel). This initial reconstruction was used to generate a reference volume using relion_image_handler (512 pixel box size, 0.8189 Å per pixel) that was subsequently used in template picking for further processing of datasets obtained on the Krios microscopes. For the vesicle datasets, particles were identified using a combination of Topaz particle picking54 and template picking in cryoSPARC, followed by several rounds of heterogeneous refinement and two-dimensional classification to deplete non-TRPM8 or obvious junk particles. After generating a consensus refinement with cryoSPARC non-uniform refinement, further classification was carried out in either RELION5 (ref. 55) or cryoSPARC. Three-dimensional classification used either a mask capturing only the channel region, or a spherical mask to limit bilayer signal. Further two-dimensional classification of three-dimensional refinements without a circular mask revealed that a range of membrane curvatures are included in reconstructions, indicating that, in our datasets, three-dimensional classes result from a distribution, rather than specific, membrane curvatures (Supplementary Fig. 1c). For the GDN-purified TRPM8 datasets, an initial reference map was generated from each dataset ab initio from particles identified with blob picker in cryoSPARC from a small sub-set of the micrographs. The ab initio model was then subjected to non-uniform refinement without symmetry and used subsequently to generate templates to process the remaining dataset. After several rounds of heterogeneous refinement, a consensus refinement was generated in cryoSPARC for further three-dimensional classification with cryoSPARC. Three-dimensional class averages were subjected to non-uniform refinement using cryoSPARC. Local resolution estimation of refined maps was carried out in cryoSPARC and visualized in UCSF ChimeraX.
Model building, pore radius and pK
a calculations
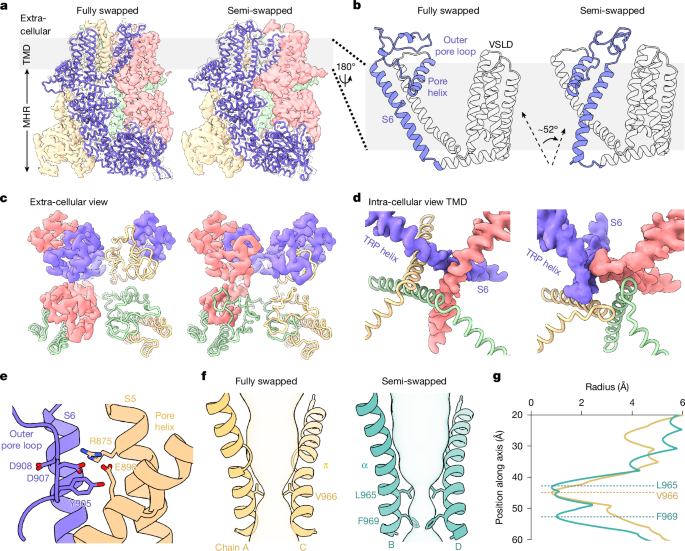
For the unliganded Pm semi-swapped structure, an initial atomic model was generated de novo using ModelAngelo (RELION v.5)56, which provided initial coordinates that defined the connectivity of the outer pore loop, S6 and TRP domain. This model was refined iteratively using Phenix real space refinement and manual refinement in COOT57 to assess density fit. Initial atomic models of Pm or Hs TRPM8 were either generated de novo or sourced from previously published structures (Extended Data Table 1). Representative densities from various regions of the corresponding reconstruction fitted with atomic models are shown in Supplementary Figs. 9–12. All models were validated using MolProbity in Phenix58. Refinement statistics of cryo-EM maps and model statistics are provided in Extended Data Table 1. A video morphing between the semi- and fully swapped models shown as a space-filling models was generated by interpolating the two PDBs (PDBs 9P90 and 9P91) using the morph command in UCSF ChimeraX (Supplementary Video 1). The HOLE program59 was used to determine all pore radii for modelled structures. For pKa calculations, models of the Pm semi-swapped closed (PDB 9P91) or fully swap closed/desensitized (PDB 9P90) structures obtained in vesicles were used as inputs for PROPKA3 (ref. 60). A summary of pKa values for ionizable amino acids found on the extracellular region is provided in Extended Data Fig. 6.
Hydrogen–deuterium exchange
Supplementary Table 3 summarizes biochemical and statistical details for HDX in this study. HDX was initiated by diluting 2 μl of 10–25 μM GDN-solubilized TRPM8 stock in an H2O buffer into 28 μl of a matching deuterated buffer to reach 93% D-content. Labelling was conducted at pH measured in deuterated solvent (pD)read 7.0 and temperatures of 4 °C, 22 °C, 30 °C or 37 °C. Labelling times were adjusted to account for the temperature dependence of intrinsic rates (kchem) based on the average kchem of the full sequence61, with details presented in Supplementary Table 3. For HDX in the presence of menthol, two conditions were employed: (1) 1 mM menthol was added to HEK293 cells expressing TRPM8 and maintained throughout purification and exchange; (2) TRPM8 was purified without menthol, and 1 mM menthol was added only to the D2O buffer. Under both conditions, HDX was performed at 22 °C. HDX time-points were collected in triplicate.
HDX was quenched at time-points ranging from 3 s to 3 days by adding 30 μl of ice-cold quench buffer (600 mM glycine, 8 M urea, 0.005% GDN, pH 2.5). Quenched samples were incubated on ice for 20 s, further diluted with 50 μl quench buffer without urea and injected immediately into a valve system maintained at 5 °C (Trajan LEAP). Non-deuterated controls and MS/MS runs for peptide assignment followed the same protocol, except that D2O buffers were replaced by H2O buffers, with immediate quenching and injection. HDX reactions were performed in random order. No peptide carryover was observed as assessed by injecting quench buffer containing 2 M urea and 0.005% GDN. In-exchange controls accounting for forward deuteration towards 41.5% D in the quenched reaction were performed by mixing D2O buffer and ice-cold quench buffer before protein addition. Maximally labelled controls (‘All D’) accounting for back-exchange were performed by incubating samples in D2O buffer for 24 h at 22 °C, followed by a 1-h heat shock at 48 °C.
Liquid chromatography–mass spectrometry
Liquid chromatography–mass spectrometry analysis for the first replicate and selected time-points for the second replicate was performed on instruments at University of California Berkeley (Marqusee laboratory), whereas the remaining time-points of replicate two and all of replicate three were analysed at UCSF (Cheng laboratory) using the same columns but different liquid chromatography–mass spectrometry instrumentation. We note that, due to differences in the liquid chromatography dead volumes, the retention times for peptides analysed at UCSF were systematically around 12 s longer than runs analysed at University of California Berkeley and, therefore, different integration bounds were used when analysing the mass spectra for those two data sets. Nonetheless, the two data sets showed high agreement in deuteration levels, as demonstrated in the repeatability assessments in Supplementary Fig. 8 and Supplementary Table 3.
Upon injection, the protein was digested online using a NepII/pepsin protease column (Affipro, catalogue no. AP-PC-006) at 10 °C. The resulting peptides were desalted by flowing across a hand-packed trap column (Thermo Scientific POROS R2 resin, catalogue no. 1112906, with IDEX C-128 1-mm internal diameter × 2-cm cartridge) at 5 °C. Digestion and desalting were completed in 2.5 min at 100 μl min−1 of 0.1% formic acid. Peptides were then separated on a C18 analytical column (Waters ACQUITY UPLC BEH C18, 130 Å, 1.7 µm, 1 mm × 50 mm, catalogue no. 186002344) with a 17-min linear gradient of 5–45% (vol/vol) acetonitrile (0.1% formic acid) delivered by a Thermo UltiMate-3000 pump (University of California Berkeley) or Vanquish Neo pump (UCSF). Eluted peptides were analysed on a Thermo Q Exactive (UC-Berkeley) or Q Exactive Plus (UCSF) mass spectrometer in positive mode (full mass spectrometry: resolution 140,000, automatic gain control target 3 × 106, maximum injection time 200 ms, scan range 300–2,000 m/z; dd-MS2: resolution 17,500, automatic gain control target 2 × 105, maximum injection time 100 ms, loop count 10, isolation window 2.0 m/z, normalized collision energy 28, dynamic exclusion 15 s).
HDX–MS data analysis and EX2 characterization
Peptides were identified using SearchGUI (v.4.0.25) and Byonic (Protein Metrics) against a search library containing TRPM8, ten additional proteins previously exposed to liquid chromatography–mass spectrometry and their reserved sequences as decoys. Search parameters included unspecific digestion, precursor and fragment tolerance of 10 ppm, precursor charge 1–8 and peptide length 5–50. HDX data were processed in HDExaminer v.3.4 (Trajan), with bimodal fitting accepted when the score exceeded the unimodal score by 0.1 or above.
The observed exchange occurred through EX2 kinetics, reporting on the equilibrium instead of opening rates of amide protons. The identification of EX2 behaviour is supported by (1) the continuous shifts in the isotopic envelopes towards higher m/z with exchange time (Supplementary Fig. 2); (2) the difference in kobs for TRPM8 labelled at various pD conditions can be attributed solely to the effect of pD on kchem (Supplementary Fig. 3). For peptides exhibiting bimodal mass envelopes, each envelope increases in mass over labelling time with exchange rates slower than kchem, and the kobs for both envelopes show pD-dependence consistent with EX2 kinetics, indicating that the two mass envelopes represent two kinetically distinct populations that each exchange through EX2 kinetics (rather than EX1 or carryover)62 (Supplementary Figs. 3 and 4). To cross-validate those bimodal fits, mass spectra were exported to HX-Express3 (ref. 63) for double-binomial fitting, with an F-test to assess the increase in fit quality over a single-binomial model. Bimodality was accepted when P < 0.01. Fitting was repeated with 20 iterations using a resampling approach in which random noise (±30% of each isotope peak intensity) was introduced in each iteration (Supplementary Figs. 5 and 6).
Each isotopic distribution was inspected manually for fit quality. Deuteration levels were adjusted for 93% D-content and back-exchange. Except for Fig. 2, for peptides displaying clear bimodality, deuteration levels represent centroids of unimodal fit to represent the overall HDX for two populations.
Quantifying thermodynamic parameters from HDX–MS
H–D exchange follows the Linderstrøm–Lang model described by the following scheme
$$\mathop{{\rm{N}}-{\rm{H}}}\limits_{\mathrm{closed}}\cdots {\rm{O}}={\rm{C}}\underset{{k}_{\mathrm{close}}}{\overset{{k}_{\mathrm{open}}}{\rightleftarrows }}\mathop{{\rm{N}}-{\rm{H}}}\limits_{\mathrm{open}}\mathop{\longrightarrow }\limits^{{k}_{\mathrm{chem}}}\mathop{{\rm{N}}-{\rm{D}}}\limits_{\mathrm{exchanged}}$$
where kopen and kclose are the opening and closing rates of hydrogen bonds for individual amides, and kchem is the chemical exchange rate of the amide in an unstructured polypeptide. At steady-state, the observed H–D exchange rate can then be described as
$${k}_{\mathrm{obs}}=\frac{{k}_{\mathrm{open}}\,{k}_{\mathrm{chem}}}{{k}_{\mathrm{open}}{+k}_{\mathrm{close}}{+k}_{\mathrm{chem}}}$$
As peptides in our dataset all exhibited EX2 kinetics (see ‘HDX–MS data analysis and EX2 characterization’), where kchem « kclose,
$${k}_{\mathrm{obs}}=\frac{{k}_{\mathrm{open}}\,{k}_{\mathrm{chem}}}{{k}_{\mathrm{open}}{+k}_{\mathrm{close}}}=\frac{{k}_{\mathrm{chem}}}{{K}_{\mathrm{eq}}+1}$$
where Keq = kclose/kopen and describes the equilibrium constant of H-bond closing and opening (folding). Folding free energy (ΔG) is then described as
$$\Delta G=-\mathrm{RTln}{K}_{\mathrm{eq}}=-\mathrm{RTln}\left(\frac{{k}_{\mathrm{chem}}}{{k}_{\mathrm{obs}}}-1\right)\approx -\mathrm{RTln}\frac{{k}_{\mathrm{chem}}}{{k}_{\mathrm{obs}}}$$
As kchem can be calculated reliably from the sequence of the peptide61, measurement of the H–D exchange rate (kobs) enables direct quantification of ΔG at peptide level. In this study, a stretched-exponential method was used to estimate ΔG from HDX as described by Hamuro37. Briefly, deuteration levels for each peptide were fit with the following equation:
$$ \% D(t)=1-\exp {[(-{kt})}^{b}]$$
where k is the exchange rate, and b is the stretch factor that describes variations of k for residues within the peptide. In those calculations, peptides with exchange rates beyond the measurement range of this study were assigned fixed values: peptides fully exchanged at the first time-point (30 s) were assigned kchem = 10 s−1, whereas peptides with no exchange at the final time-point (30 h) were assigned kobs = 10−5 s−1. Changes in folding free energy (ΔΔG) under two conditions (with and without menthol) were then calculated based on the following equation:
$$\Delta \Delta G=-{\rm{l}}{\rm{n}}(10){\rm{R}}{\rm{T}}\left[-\frac{\gamma }{{\rm{l}}{\rm{n}}(10)}\left(\frac{1}{{B}_{1}}-\frac{1}{{B}_{2}}\right)-{\log }_{10}\frac{{k}_{1}}{{k}_{2}}\right]$$
where \(\gamma \) is the Euler–Mascheroni constant (approximately 0.577215665). For Fig. 4a and Extended Data Fig. 5a, an average difference of deuteration levels was used to estimate the ΔΔG at temperatures relative to 22 °C, as described by Hamuro37, as some regions exhibited incomplete exchange within our labelling times, which resulted in low confidence in fitting results with the stretched-exponential method. In this strategy, ΔΔG was calculated based on the following equation:
$${\rm{\Delta \Delta }}G=-\text{ln}(10){\rm{RT}}\frac{W}{p}\mathop{\sum }\limits_{t=1}^{p}( \% {D}_{1,t}- \% {D}_{2,t})$$
where p is the number of time-points and t is a specific time-point. For each temperature window, van’t Hoff analysis was employed to determine ∆H°, entropy contribution (TΔS°) and Gibbs free energy (ΔG°) extrapolated to 25 °C, assuming no change in Cp, based on the following equation:
$$\mathrm{ln}{K}_{\mathrm{eq}}=-\frac{\Delta H^\circ }{R}\frac{1}{T}+\frac{\Delta S^\circ }{R}$$
In both quantification strategies, residue-level ΔΔG and ∆H° values were determined from a weighted average of peptide-level parameters, excluding the first two N-terminal residues due to rapid back-exchange. We note that not all peptides identified exhibited complete exchange within our labelling times and, therefore, the thermodynamic parameters quantified here represent the best approximation within the scope of the study. Nonetheless, our approach effectively captures relative energetic changes associated with menthol-binding and temperature changes that are consistent with our structural and functional studies.
Fura-2-AM calcium imaging and data analysis
Calcium imaging experiments were carried using Fura-2-AM as a ratiometric fluorescent indicator (as described previously64) with temperature monitoring throughout imaging. Briefly, adherent HEK293T cells grown at 37 °C with 5% ambient CO2 were seeded in a 12-well plate (1 ml volume) to a final density of 30–40% confluence and transfected with 0.5 mg ml−1 plasmid using Lipofectamine 3000 (ThermoFisher Scientific) according to the manufacturer. Cells were allowed to transfect for 12–14 h. Borosilicate cover slips (12 mm, Bellco Glass) were incubated with a 1:100 solution of Matrigel matrix (Corning) prepared in serum-free DMEM (Gibco) for 1 h, then washed once with DMEM and left in a 24-well plate with DMEM supplemented with 10% bovine calf serum. Transfected cells were dissociated gently and replated directly onto coverslips and allowed to attach for 2–3 h before imaging experiments were carried out. Attached cells were washed once with Ringer’s solution (10 mM HEPES-Tris pH 7.40, 140 mM NaCl, 5 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 10 mM glucose; 290–310 mmol kg−1), and then incubated for 0.5 h at room temperature in the dark in Ringer’s solution supplemented with 10 μg ml−1 Fura-2-AM (ThermoFisher Scientific) and 0.02% Pluronic F127. After incubation, cells were washed once with Ringer’s solution and allowed to incubate further in Ringer’s solution without Fura-2-AM or Pluronic F127 for 0.5 h. Dye-loaded cells were then imaged using an inverted microscope with 340 and 380 nm excitation (Sutter, Lambda LS Illuminator). The temperature for each experiment was held at 32–37 °C by perfusing Ringer’s solution through a Peltier device controlled with a heating module (Warner TC-324B) calibrated to hold the desired temperature continuously. Cells were held at this temperature range for 5–10 min before each recording. Bath exchange into low temperatures was achieved by perfusing Ringer’s solution cooled with ice water into the recording chamber. Temperature was monitored and synchronized using a thermistor reading input to the heating module that was digitized through an Axon Digidata 1550B module and monitored with pCLAMP10. Recordings were obtained as stacked videos of individual 340/380 frames with a 1-s interval in Micro-Manager65. Videos of ratio images were calculated with an ImageJ macro script according to known procedures66. Peak amplitudes of calcium signals corresponding to either cold or menthol, were obtained and normalized to maximum calcium signal obtained using 10 µM ionomycin before further statistical analyses.
Sequence alignment of TRPM8 orthologues
Sequences of avian or mammalian TRPM8 were obtained from OrthoDB v.12.2 (orthodb.org) by curating the phylogenetic clades of Sauropsida (which includes avian species), and Mammalia, respectively. Multiple sequence alignments were generated for Sauropsida (450 sequences) and Mammalia (390 sequences) using Clustal Omega (https://www.ebi.ac.uk/jdispatcher/msa/clustalo). The resulting outputs were used to plot individual sequence logo representations of the avian or mammalian TRPM8 orthologues near Y905 (for Pm TRPM8) or V915 (for Hs TRPM8) using R (ggseqlogo)67.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
First Appeared on
Source link

