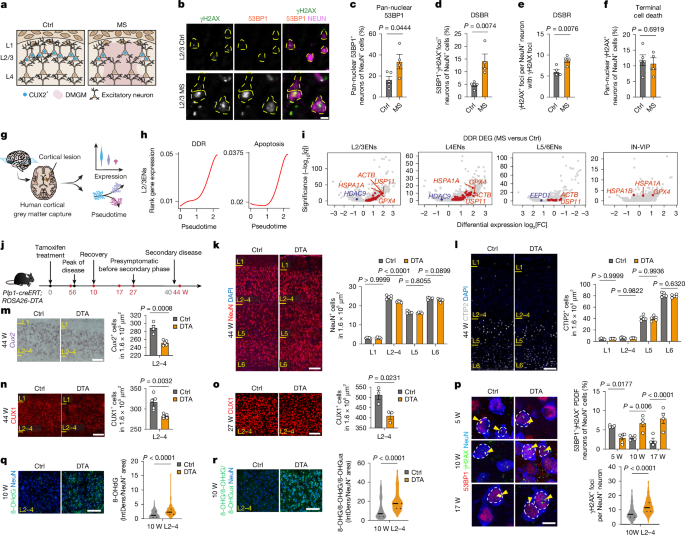
DNA damage burden causes selective CUX2 neuron loss in neuroinflammation
Human tissue sample collection and compliance
Single-nucleus capture and RNA-seq analysis of age- and sex-matched human control (9) and MS (12) cortices was performed previously2. For this study, human post-mortem brain samples with MS grey matter pathology were sourced from the UK Multiple Sclerosis Tissue Bank at Imperial College London. Ethical approval for the use of these samples was granted by the National Research Ethics Committee in the UK (08/MRE09/31). In total, nine snap-frozen brain blocks (four from patients with MS and five from individuals without diagnosed neurological disease) were analysed using immunohistochemistry. For CUX2 and ATF4 expression analyses, the 25-year-old sample was obtained from University of California, San Francisco’s Pediatric Neuropathology Research Laboratory and collected in accordance with guidelines established by the Committee on Human Research (UCSF) and approved by the Institutional Review Board. Participant details are provided in Supplementary Table 1.
Primary rat glial cultures
Fresh Wistar/Han postnatal day 5 rat heads were obtained from Charles River Laboratory (UK) in ice-cold Hibernate-A medium (Thermo Fisher Scientific, A1247501). Whole brains were extracted, homogenized and subjected to centrifugation (3 min, 100g at room temperature). The supernatant was removed and the tissue was resuspended in a dissociation solution (34 U ml−1 papain (Lorne, LS003126) and 20 μg ml−1 DNase type I (Millipore, 10104159001) in Hibernate-A medium) that had been heat-activated at 37 °C and shaken for 20–30 min at 55 rpm and 37 °C. The tissue was centrifuged for 5 min at 200g, the supernatant was aspirated and then the tissue was resuspended in a cold neutralizing solution (Hibernate A medium supplemented with 1× B27 (Thermo Fisher Scientific, 17504044) and 2 mM sodium pyruvate (Sigma-Aldrich, S8636)). Tissue was triturated using a serological pipette ten times and, after 1 min rest, the cell suspension (supernatant) was transferred through a 70-μm cell strainer into isotonic Percoll (90%, Sigma-Aldrich, GE17-5445-01). The remaining tissue aggregates were subjected to a further three rounds of trituration and 1 min resting, and the supernatant was transferred into isotonic Percoll. The cell suspensions were brought to a final Percoll concentration of 22.5% by adding DMEM/F12 with HEPES (Thermo Fisher Scientific, 11-039-021) and centrifuged for 20 min at 800g. The supernatant was removed, and the brain cell pellet (bottom 3 ml) was resuspended in HBSS without calcium and magnesium (Thermo Fisher Scientific, 14170112), then centrifuged for a further 5 min at 300g; the supernatant was next removed and the cells were resuspended in 1 ml of red blood cell lysing buffer (Sigma-Aldrich, R7757). The suspension was incubated for 90–120 s before washing with HBSS without calcium and magnesium. The cells were centrifuged for 5 min at 300g before resuspending in 500 μl sorting solution (1× PBS supplemented with 2 mM EDTA (Thermo Fisher Scientific, 15575020), 2 mM sodium pyruvate, 0.5% BSA and 25 μg ml−1 insulin (Sigma Aldrich, I9278)). The cells were counted and 2 μl of CD11b/c rat microbeads (Miltenyi Biotec, 130-105-634) was added per 1 million cells and incubated for 15 min at 4 °C with gentle agitation on a tube roller. A further 8 ml of sorting solution was added before the cell/bead mix was centrifuged for 5 min at 300g. The supernatant was removed, and cells were resuspended in 2 ml sorting solution. Cells were added to an LS column (Miltenyi Biotec, 130-042-401) fitted to a QuadroMACS Separator (Miltenyi Biotec, 130-091-051) stand that had been pre-wet with sorting solution. The column was washed three times with 2.5 ml cold sorting solution. Microglia were collected from the column by disassociating the column from the stand, adding 2 ml microglial medium (DMEM/F12 supplemented with 60 μg ml−1 N-acetyl cysteine (Sigma-Aldrich, A9165), 10 μg ml−1 insulin, 1 mM sodium pyruvate, 1× SATO (100× SATO: 1.61 mg ml−1 putrescine dihydrochloride (Sigma-Aldrich, P5780), 4 µg ml−1 sodium selenite (Sigma-Aldrich, S526110), 60 µg ml−1 progesterone (Sigma-Aldrich, P8783), 41.25 mg ml−1 BSA (Sigma-Aldrich, A4919), 5 mg ml−1 apo-transferrin (Sigma-Aldrich, T1147) in DMEM/F12 (Gibco, 11039021)) before using a plunger to gently release the cells. Microglia were counted and seeded at 400,000 cells per well on 12-well plates precoated with 5 μg ml−1 poly-d-lysine (Sigma-Aldrich, P6407). To obtain astrocytes, the cell flow-through was further sorted to remove oligodendroglia. Flow-through suspensions were centrifuged for 5 min at 300g and the supernatant was aspirated. Cells were resuspended in 500 μl of sorting solution before adding 1.7 μl of mouse IgM anti-A2B5 antibody (about 1:250, Sigma-Aldrich, MAB312, AB-94709) and incubated for 25 min at 4 °C with gentle agitation. The cell suspension was diluted with sorting solution and centrifuged for 5 min at 300g. The supernatant was aspirated, and the pellet was resuspended in 160 μl of sorting solution with 40 μl of rat anti-mouse IgM microbeads (Miltenyi Biotec, 130-047-302) and further incubated for 15 min at 4 °C, with gentle agitation at the 5- and 10-min mark. The cells were sorted through the LS column as described above for microglia but the flow-through was collected for further astroglial isolation. The solution was centrifuged for 5 min at 300g and then resuspended in astrocyte medium (DMEM/F12 without L-glutamine (Thermo Fisher Scientific, 21331020) supplemented with 1 mM sodium pyruvate, 60 μg ml−1 N-acetyl cysteine, 1× N2 (Thermo Fisher Scientific, 17502-001), 1× B27 (Gibco, 12587-010), 1% penicillin–streptomycin, 10 μg ml−1 insulin and 10 ng ml−1 human recombinant HB-EGF (Peprotech, 100-47-100UG)). The cell solution was added to a flask coated with 5 μg ml−1 poly-d-lysine and glia were allowed to attach for 1 h at 37 °C. After 1 h, the unattached cells (predominantly containing neurons and endothelia) were washed away, and fresh astrocyte medium was provided. After 24 h, astrocytes were passaged by first washing with PBS minus calcium and magnesium chloride (Sigma-Aldrich, D8537) before replacing with trypsin-EDTA (0.05%; Gibco, 25300054) at 37 °C for 5 min. Astrocyte medium was added and astroglia were collected and centrifuged at 300g for 5 min. Astrocytes were resuspended in astrocyte medium and plated at 300,000 cells per well of 12-well plates. Cells were then subjected to rat IFNγ (Thermo Fisher Scientific, 400-20) before collection.
Demyelination and neuroinflammation models
ROSA26–eGFP-DTA mice52 were bred to the PLP/creERT transgenic mice53 to generate the PLP/creERT;ROSA26–eGFP-DTA (DTA) mice, as previously described17,18. All mice were on the C57BL/6J background. PLP/creERT;ROSA26–eGFP-DTA (DTA) mice and ROSA26–eGFP-DTA littermates (control; aged 5–7 weeks) were injected intraperitoneally with 0.8 mg of 4-hydroxytamoxifen (Hello Bio, HB6040) per day, either for 4 consecutive days (males) or 3 consecutive days (females). Brain tissue was collected from the DTA and control animals at the indicated timepoints after tamoxifen injection. Equal numbers of male and female mice were used for experiments, which were conducted in compliance with Northwestern University’s Animal Care and Use Committee (IACUC) guidelines.
Myrffl/fl were crossed to Sox10creERT mice to generate Myrf-cKO mice as previously described20. All of the mice were on a mixed C57BL/6N and C57BL/6J background and creERT-negative littermates served as non-demyelinated controls. Eight-week-old mice were injected intraperitoneally with 100 mg per kg tamoxifen (T5648, Sigma-Aldrich) dissolved in corn oil (C8267, Sigma-Aldrich) for 5 days. These experiments were conducted in compliance with the Institutional Animal Care and Use Committee of OHSU.
Ectopic IFNγ expression model
Gfap-tTA mice on the C57BL/6J background were crossed with TRE-Ifng mice on the C57BL/6J background to generate Gfap-tTA;TRE-Ifng double-transgenic mice, as described previously54,55. To suppress IFNγ transcription, doxycycline (200 ppm) was administered in the diet (Envigo) from conception. Mice were maintained on doxycycline throughout development, and doxycycline repression was lifted at 8 weeks of age. Brain tissue was collected from both double-transgenic and single-transgenic control mice at 12 months after doxycycline withdrawal. These experiments were conducted in compliance with Northwestern University’s Animal Care and Use Committee (IACUC) guidelines.
KO and cKO lines
Cux2cre56,57, Cux2creER57, Atf4fl58 and WT mice were handled according to guidelines set by the University of California, San Francisco and housed within a barrier facility on a 12 h–12 h light–dark cycle. Mice were housed with up to four other same-sex cage mates in standard rodent cages and provided food and water ad libitum. Both male and female mice were used for all experiments. All animal protocols and procedures were approved by UCSF’s Institutional Animal Care and Use Committee (IACUC).
Neonatal hypoxia and acute neuroinflammation model
Neonatal mice, along with their parents, were exposed to chronic hypoxia by receiving 10% fractional inspired oxygen from P3 to P14. At P7, LPS (Sigma-Aldrich, L2630, 93572-42-0) was administered through intraperitoneal injection at a dose of 2 mg per kg. After P14, both pups and parents were returned to normoxic conditions until brain tissue was collected at P28. Mice were monitored daily. The oxygen concentration was continuously monitored and maintained using an oxygen modulator (Biospherix, ProOx110). This procedure was approved by UCSF’s Institutional Animal Care and Use Committee (IACUC).
Acute demyelination with cuprizone
Tamoxifen (T-5648, Sigma-Aldrich, 10540-29-1) was dissolved in a solution of 90% corn oil and 10% ethanol and administered intraperitoneally to Cux2creERAtf4fl mice starting at 8 weeks of age. Each mouse received 2 mg of tamoxifen per day for 5 consecutive days, followed by a 2 week rest period.
Next, the same mice were fed a diet containing 0.3% (w/w) cuprizone (Sigma-Aldrich, C9012, 370-81-0) mixed into standard chow. Cuprizone was administered continuously for 6 weeks to induce robust demyelination. Mice were monitored daily and provided with fresh cuprizone-containing food until brain tissue was collected at the end of the 6-week period. This procedure was approved by UCSF’s Institutional Animal Care and Use Committee (IACUC).
Collection of tissue for histology
For histology, mice were transcardial perfused with 4% paraformaldehyde (PFA), and brain tissue was collected. The brains were post-fixed overnight in 4% PFA and washed with PBS before being embedded in OCT. Then, 16-μm sections were obtained on Superfrost Plus slides using a Leica Cryostat. The sections were stored at below −70 °C.
Collection of tissue for snRNA-seq
For snRNA-seq, cortices were collected in ice-cold DEPC-PBS under RNase-free conditions, frozen with dry ice and stored at −80 °C until sequencing was performed.
For snRNA-seq on DTA mice, 1 mm coronal sections were collected using a stainless-steel brain matrix. From these sections, 0.75 mm punches were collected from the corpus callosum and cortex, which were frozen and shipped to UCLA, where the sequencing was performed.
snRNA-seq
For snRNA-seq, mRNAs from isolated nuclei were barcoded using the 10x Genomics 3′ Gene Expression v3.1 kit. Libraries were sequenced on the NovaSeq 6000 system using an S4 flow cell. Raw data were processed with CellRanger (v.7.0.1) using the mm10-2020-A mouse genome reference for embryonic samples or CellRanger (v.8.0.1) with GRCm39-2024-A mouse reference for postnatal samples. Ambient RNA contamination was removed using CellBender (embryonic, v.0.1.0; postnatal, v.0.3.0).
Cell type annotation
Cell type annotation was performed using a combination of clustering and reference-based approaches. We used the Deep Embedding for Single-cell Clustering (DESC, v.2.1.1)59 package for dimension deduction, batch normalization and clustering, using the top 2,048 high DEGs (scanpy60, v.1.8.1) and a three-layer encoder network (1,024, 256, 32) for feature extraction. Clusters were annotated using Pegasus (v.1.8.1) for automated cell type suggestions, with further validation against the Allen Brain Atlas and the Linnarsson Atlas for cortical regions61,62. For the integrated Popko dataset, a hierarchical annotation strategy was applied, sequentially removing non-neuron cells, non-cortex neurons and inhibitory neurons, with manual marker gene verification at each step.
DEG analyses
DEGs for Cux2creAtf4fl mice were determined in Omics playground63 (v.2.8.19) by performing t-tests (standard, Welch) and limma (no trend, trend, voom), edgeR (QLF, LRT) and DESeq2 (Wald, LRT) tests and taking the highest q value for tests with cutoffs of a false-discovery rate (FDR) of 0.05 and a log2-transformed FC of 0.1.
DEGs for DTA mice were determined instead in Python using Scanpy’s rank_genes_groups function and using Wilcoxon rank-sum tests to determine statistical significance.
Volcano plots were generated using the Hiplot (ORG) (https://hiplot.org) Volcano plot APP (v.0.1.0) using a Creative Commons Attribution 4.0 International Licence to visualize DEGs with highlighted genes. Moreover, DEG distribution box plots were created by randomly sampling 1,000 cells from each cluster, repeated 100 times. The filtered number of DEGs (P < 0.05 and log2[FC] > 0.1) in each cluster was recorded as a .csv file and then visualized using ggplot2 v.3.5.1. Minor visualization modifications were made to plots in Adobe Illustrator such as removing unwanted lines and text formatting.
Trajectory pseudotime and real-time analysis
Pseudotime analysis was based on previously established trajectories from our previous sequencing study2. Gene expression dynamics over pseudotime and real time were analysed by calculating the mean gene expression or gene list scores at each timepoint, followed by polynomial fitting using numpy. For pseudotime analysis, four timepoints were estimated based on the region where the cells are from (healthy controls, normal appearing white matter, acute lesion and chronic lesion region), while for real-time analysis, the timepoints are based on the age of the animals when collected. Gene list scores were calculated using Scanpy’s score_genes or Seurat’s AddModuleScore function, with 500 random genes selected as negative controls to establish baseline scores.
Interactive cell annotation and analyses were performed using Cellxgene-VIP (v.3.0) and Omics playground.
GO analysis
DNA repair GO analysis (Supplementary Table 3) was performed in g.profiler (v.4.2.8)64 (https://biit.cs.ut.ee/gprofiler/gost) with statistical enrichment calculated using the g:SCS test with experiment-wide threshold of α = 0.05.
GO analysis on DEGs was conducted and visualized using the ClusterProfiler65 (v.4.14.6) package in R. DEGs identified by Scanpy’s rank_gene_groups function (P < 0.05, log2[FC] > 0.2) were used as input. Enrichment significance was assessed using thresholds of P < 0.05 and q < 0.05, and redundant GO terms were reduced using the simplify() function.
DDR gene list
We compiled the list of 698 genes considered as DDR genes by combining GOterms GO:0006281DNArepair and HALLMARK DNA_REPAIR (M5898), and consolidating with previously curated human DNA repair genes by R. Wood and M. Lowery (https://www.mdanderson.org/documents/Labs/Wood-Laboratory/human-dna-repair-genes.html)66,67,68.
Quantitative PCR
RNA was isolated from cells in culture by first washing with 1× PBS and then replacing PBS with TRI reagent (Cambridge Bioscience, R2051) for 5–10 min at room temperature. RNA was isolated using the Direct-zol RNA miniprep kit (Cambridge Bioscience, R2051) according to the manufacturer’s instructions. cDNA was generated using the Zymoscript RT Premix kit (Cambridge Bioscience, R3012). cDNA (approximately 20–200 ng diluted in H2O) was amplified in a mastermix containing 0.1 μM forward and reverse primers (Supplementary Table 12) and PowerUp SYBR Green Master Mix (1×, Applied Biosystems, Thermo Fisher Scientific, A25777). Quantitative comparative PCR was performed on the Applied biosciences Quantstudio 12K Flex Real-time PCR system (Thermo Fisher Scientific, 4470661) using the default SYBR comparative CT program.
Neurogenin-2 knock-in iPS cells
Before nucleofection, the ribonucleoprotein complex was assembled and incubated at room temperature. Twenty μg of Cas9 (Integrated DNA Technologies, 1081061) was complexed with 150 pmol of the synthetic gRNA targeting the AAVS1 locus in nuclease-free duplex buffer (Integrated DNA Technologies, 11-01-03-01). After 30 min incubation, 2 μg of the donor plasmid pUCM-AAVS1-TO-hNGN2-T2A-Zeo was added and mixed with 106 cells (iPS cells, CAMi014-A (corrected line))69 that were then nucleofected in 100 μl cuvettes using the Amaxa 4D-Nucleofector (Lonza) and the P3 Primary Cell 4D-Nucleofector X Kit (Lonza, V4XP-3024) using the CA-137 program. Cells were then plated with 4 μM Cas9 Electroporation Enhancer (Integrated DNA Technologies, 1075916) and 10 μM of ROCKi. The medium was refreshed after 24 h.
Then, 3 days after nucleofection, the cells were pooled and assessed for mCherry expression by flow cytometry using the CytoFLEX (Beckman Coulter). By plotting SSC-H-Lin versus FSC-H-Lin live cells, the population was revealed (centre) and gated. The single-cell population was isolated by plotting the live cell subpopulation on an FSC-H-Lin versus FSC-ALin. Single cells were analysed for mCherry expression by plotting FSC-H-Lin versus PE-A-Log; mCherry positive population was gated to the right of the 104 mark on the PE-A axis. Flow cytometry data were analysed using FlowJo v.10.7.1 (Becton Dickinson). Simultaneously, cells were observed for mCherry expression on the EVOS fluorescence microscope. Recombinant cells were selected with 1 μg ml−1 puromycin for 5 days.
After puromycin selection, clones were isolated by plating 103 filtered cells with a 30 μm mesh in a 10 cm dish and picked when colonies were visible by eye. Colonies were expanded and selected based on the ability to differentiate into NGN2-iNs.
Cell lines and culture
SHSY5Y neuroblastoma cells were obtained from ATCC (CRL-2266) and maintained in DMEM/F-12 (Sigma-Aldrich, D8437) supplemented with 10% FBS and penicillin–streptomycin (LifeTech/Gibco, 15140-122; neuroblastoma medium) and cryofrozen with the addition of 10% DMSO (Sigma-Aldrich, D2438). Cells were passaged by washing once with PBS minus calcium chloride and magnesium chloride (Sigma-Aldrich, D8537) and replacing with trypsin-EDTA (0.05%; Gibco, 25300054) at 37 °C for 2 min. Cells were collected in neuroblastoma medium and pelleted by centrifugation at 300g for 5 min. Cell pellets were resuspended in neuroblastoma medium.
NGN2-iPS cells were maintained in Stemflex medium (Thermo Fisher Scientific, A3349401) on six-well plates coated with Corning Matrigel growth factor reduced basement membrane matrix (Corning, Scientific Laboratories Supplies, 356230). Cells were passaged by washing once with PBS minus calcium chloride and magnesium chloride (Sigma-Aldrich, D8537) and incubating in 0.5 mM EDTA in 1× PBS (Thermo Fisher Scientific, 15575020) for 5 min at 37 °C. Cells were collected in Stemflex and pelleted by centrifugation at 300g for 5 min. Cell pellets were resuspended in Stemflex supplemented with 10 μM ROCK inhibitor Y-27632 (Abcam, ab120129) and either plated for further growth or cryofrozen by addition of 10% DMSO.
hTERT RPE-1 cells were obtained from ATCC (CRL-4000) and maintained in RPE-1 medium: DMEM/F12 high glucose (Thermo Fisher Scientific, 31053028) supplemented with 10% FBS, 1× MEM-NEAA (Life Technologies, 11140035) and 1× GlutaMax (Gibco, 31331028). Cells were passaged as described for SHSY5Y.
All of the cell lines tested negative for routine Mycoplasma testing by PCR and rapid test. No cell lines were authenticated.
Induced neuron generation and culture
NGN2-iPS cells were induced to become neurons as follows: NGN2-iPS cells were dissociated into single cells using Accutase (in place of EDTA, Life Technologies, A1110501) at 60–80% confluence and 250,000 cells per well were plated in six-well culture dishes coated with Corning Matrigel growth-factor-reduced basement membrane matrix. On day 1 and 2, the medium was replaced with fresh D1-2 differentiation medium (DMEM/F12, GlutaMax (Gibco, 10565-018) supplemented with 1× MEM-NEAA (Life Technologies, 11140035), 1% penicillin–streptomycin, 1× N2 (Thermo Fisher Scientific, 17502-048), 1 μg ml−1 doxycycline (Sigma-Aldrich, D9891, 24390-14-5) and 55 μM 2-mercaptoethanol (Gibco, 21985023)). On day 3, the medium was replaced with D3+ differentiation medium (Neurobasal (Gibco, 21103-049), supplemented with 1× B27 (Gibco, 12587-010), 1% penicillin–streptomycin, 1× GlutaMax, 55 μM 2-mercaptoethanol, 1 μg ml−1 doxycycline, 10 ng ml−1 NT3 (Peprotech, 450-03) and 10 ng ml−1 BDNF (Peprotech, 450-02)). On day 4, 96-well plates were coated with poly-l-lysine (0.1 mg ml−1, Sigma-Aldrich, P1524) for 1 h at 37 °C. Induced neuron cultures were washed once with PBS minus calcium chloride and magnesium chloride and Accutase was added for 5 min at 37 °C. Cells were collected in 1× PBS and pelleted by centrifugation for 5 min at 300g. Cells were resuspended in D3+ medium supplemented with 10 μM ROCK inhibitor Y-27632. Cells were plated at 100,000 cells per well of a 96-well or 24-well plate. On day 5 and 6, medium was refreshed with D3+ differentiation medium. From day 8, cells were maintained by replacing half of the medium with D3+ differentiation medium minus doxycycline every other day.
Plasmid construction, lentivirus preparation and infection
Lentivirus donor DNA plasmids were ordered from Vectorbuilder and were as follows: pLV[Exp]-Puro-CBh>hCUX2(NM_015267.4)/GST/T2A/eGFP (CUX2-OE), pLV[Exp]-Puro-CBh>hATF4(NM_001675.4)*/Flag/T2A/mCherry (ATF4-OE) or pLV[Exp]-Puro-CBh>eGFP (GFP-OE). The specific constructs used for hTERT RPE-1 cell overexpression were as follows: pLV[Exp]-Puro-CBh>hATF4(NM_001675.4]/3×GS/Halo, pLV[Exp]-Puro-CBh>hCUX2(NM_015267.4)*/3×GS/Halo and pLV[Exp]-Puro-CBh>hRPA3(NM_002947.5)/HA/T2A/eGFP.
HEK293T cells were sourced from ATCC (CRL-3216) and maintained as described for the neuroblastoma cell line but were cultured without antibiotics. To generate lentivirus, HEK293T cells were grown in 15 cm dishes and split 1:1 at 70% confluence 1 day before transfection. Then, 4.5 μg of psPAX2 (Addgene, 12260) and 4.5 μg of vSVG (Addgene, 14888) were combined with 4.5 μg of donor DNA in 2.25 ml Opti-MEM reduced serum medium. Next, 54 μl of Lipofectamine 2000 Transfection Reagent (Thermo Fisher Scientific, 11668030) was combined with 2.25 ml Opti-MEM reduced serum and then mixed with the DNA/Opti-MEM solution and incubated at room temperature for 20 min. DNA–lipid complexes were added to each 15 cm plate and incubated for 15 h in the incubator. The medium was changed to target cell medium (for example, neuroblastoma medium) and the supernatant was collected after a further 24 h, refreshed and then collected again after another 24 h. The collected supernatants were spun at 500g for 10 min to remove cell debris and filtered through 0.45 µm filter before being aliquoted and stored colder than −70 °C until use.
Lentiviral transduction was performed by combining lentivirus-containing supernatants 1:1 with fresh complete medium (neuroblastoma or D3+) and applying to neuroblastoma or NGN2-iNs in 24-well or 6-well plates. Spinoculation was performed at 25–35 °C by centrifugation at 1,000g for 30 min to 1 h. The medium was changed the next day for NGN2-iNs or after 48 h for neuroblastoma. Lentiviral-transduced cell lines were established by selecting with puromycin (10 µg ml−1; Thermo Fisher Scientific, J67236.XF) from day 3 after transduction.
Comet assays
Neuroblastoma cells were plated at 500,000 cells per well in six-well plates. The next day, cells were exposed to 200 μM TBHP (Sigma-Aldrich, 458139, 75-91-2) or left untreated for 1 h. Treated cells were recovered for 0–4 h before collecting cells in 1× PBS by scraping. Cell suspensions were then combined 1:4 with low-melting-point Comet agarose (R&D systems, 4250-050-02) and 75 μl was applied to a single well of a Comet slide (R&D systems, 4250-200-03). The slides were set for 15 min at 4 °C, before being subjected to lysis solution (R&D systems, 4250-050-01) for 1 h at 4 °C. Alkaline unwinding was performed at room temperature for 20 min by submerging the slides in 300 mM NaOH, 1 mM EDTA, in distilled H2O. The slides were transferred to an electrophoresis chamber submerged in alkaline unwinding solution and run at 1 V cm−1 for 20 min (where cm = distance between electrodes). After electrophoresis, slides were submerged in 70% (w/v) ethanol in distilled H2O for 5 min and dried overnight at room temperature. Cells were stained by applying SYBR green stain diluted in 1 mM EDTA and 10 mM Tris-HCl (Thermo Fisher Scientific, 10573145). Images were collected on the EVOS FL upright microscope.
MTT viability assay
Neuroblastoma cells were plated at 40,000–50,000 cells per well in 96-well plates. NGN2-iNs were plated at 100,000 cells per well on day 4. On day 5 (neuroblastoma) or day 19 (NGN2-iNs), a mastermix containing DNA-damaging agents (TBHP; Carboplatin, Sigma-Aldrich, C2538, 41575-94-4; 6-thioguanine, Sigma-Aldrich, A4882, 154-42-7; thapsigargin, Abcam, AB120286, 67526-95-8; topotecan, Apex Bio, B2296, 119413-54-6; colchicine, Sigma-Aldrich, C975, 64-86-8; etoposide, Sigma-Aldrich, 341205, 33419-42-0) diluted in fresh medium was made, and 100 μl was delivered to each well. Then, 24 h later, MTT reagent (Abcam, ab211091) was mixed 1:1 in DMEM (Thermo Fisher Scientific, 11-039-021) and added to cells for 45 min to 1 h at 37 °C and 5% CO2. MTT solution was removed and replaced with pure methanol (100 μl per well of a 96-well plate). Colorimetric cell viability was measured on a Spectrostar nano (BMG Labtech) plate reader using Spectrostar nano firmware (v.1.11) and software (v.2.12) at 570 nm and 690 nm.
CellROX probe assay
Primary rat astrocytes, microglia or human NGN2-iNs were pretreated with IFNγ (Thermo Fisher Scientific, human: PHC4031; rat: 400-20), and supplemented with 10 μM NAC (Sigma-Aldrich, A9165-5G) or 1 μM Mito-TEMPO (Sigma-Aldrich, SML0737). CellROX reagent (5 μM; Invitrogen, C10448) was added to the medium and cells were returned to the 37 °C incubator for 30 min. Images were collected on an EVOS FL upright microscope and analysed for fluorescence using a custom CellProfiler pipeline (v.4.2.8; available at GitHub: https://github.com/RowitchLab/Code_for_Cux2_Atf4_paper).
Luciferase assay
The human RPA3 promoter (1,500 bp upstream to 100 bp downstream of the transcriptional start site) was cloned into the pGL4.10[luc2] vector (Promega, E6651) using Kpn1 digestion and Infusion cloning (Takara, 638945) with specific primers (Supplementary Table 12). The following vectors derived from Vectorbuilder were used to express GFP and CUX2: pRP-CBH-ORFstuff-eGFP and pRP-CBH-hCUX2(NM_015267.4)-eGFP.
In total, 100,000 HEK293T cells were plated in 96-well dishes and transfected the next day with pGL4.10[luc2] or pGL4.10[Rpa3-luc2] (325 ng per well), overexpression vectors (9.55 fmol) and pGL4.75[hRluc/CMV] (5 ng per well) vectors using Lipofectamine 2000 Transfection Reagent. Then, 24 h later, a dual-luciferase reporter assay (Promega, E1910) was performed according to the manufacturer’s instructions and firefly luciferase and Renilla luciferase activity was measured on the Promega Glomax luminometer. Firefly luciferase readings were normalized to Renilla luciferase as an internal transfection control.
NHEJ reporter assay
Non-blunt NHEJ reporter plasmids were a gift from E. Rajendra (Artios Pharma) and prepared by I-SceI digestion and purification as described previously70.
A total of 200,000 hTERT RPE-1 cells was transfected with siRNA (Supplementary Table 12) using Lipofectamine RNAiMAX (Thermo Fisher Scientific, 13778100) at passage according to the manufacturer’s instructions and plated immediately in six-well dishes. After 72 h, cells were passaged by washing once with PBS minus calcium chloride and magnesium chloride and replacing with trypsin-EDTA (0.05%; Gibco, 25300054) at 37 °C for 2 min. Cells were collected in RPE-1 medium and pelleted by centrifugation at 300g for 5 min. Cell pellets were resuspended in RPE1 medium and counted. Meanwhile, non-blunt NHEJ substrate (0.5 μg DNA per 1 × 106 cells) was combined with the pGL4.50[luc2/CMV/Hygro] Vector (Promega, E1310; 0.66 μg DNA per 1 × 106 cells) in JetPRIME buffer (Sartorius, 201000003). Diluted DNA was then complexed with JetPRIME reagent (2.34 μl reagent per 1 × 106 cells, Sartorius, 101000046) and incubated for 10 min at room temperature. Cells were then combined with DNA transfection complexes and plated at 27,000 cells per well in 96-well plates. Then, 24 h later, cells were washed with PBS, lysed with 20 μl of 1× passive lysis buffer (Promega E1941) for 15 min at room temperature. The Nano-Glo Dual-Luciferase Reporter Assay (Promega, N1610) was performed according to the manufacturer’s instructions. Firefly and NanoLuc luciferase were detected using the Promega Glomax luminometer. NanoLuc measurements (NHEJ repaired substrate) were normalized to the Firefly measurement to account for variability in transfection efficiency. Readings from 10 ×100,000 plated cells were averaged per trial.
Immunohistochemistry analysis of rodent brain tissue
Cryosections were removed from the freezer and air dried for 15 min at room temperature. Slides were washed for 10 min in 1× PBS before antigen retrieval was performed either (1) in a steam pot for 5 min immersed in sodium citrate buffer (10 mM sodium citrate, 0.05% Tween-20, pH 6.0); or (2) in an 80 °C water bath immersed in 1× citrate buffer (Sigma-Aldrich, C9999) for 30–60 min. The slides were washed in 1× PBS for 10 min twice, before blocking solution was added for 1 h (10% normal donkey or goat serum in 0.1% Triton X-100/1× PBS). Primary antibodies (Supplementary Table 11) were added to blocking solution and incubated on slides overnight at 4 °C or room temperature. The slides were washed in 1× PBS for 10 min thrice. Secondary antibodies (Supplementary Table 11) were diluted in 0.1% Triton X-100/1× PBS and incubated for 1 h on slides. The slides were washed three times in 1× PBS for 10 min each, before DAPI (0.25 μg ml−1, Sigma-Aldrich) or Hoechst (1:1000, Life Technologies, H3570) was added for 20 min at room temperature. Slides were coverslipped in ProLong Gold Antifade Mountant (P36930, Thermo Fisher Scientific).
In situ hybridization on rodent brain tissue
For in situ hybridization staining, tissue sections were incubated overnight at 65 °C with a diluted, denatured DIG-labelled antisense mouse Cux2 probe. This was followed by three post-hybridization washes at 65 °C and two additional washes at room temperature. Next, the sections were incubated overnight at 4 °C with an anti-digoxigenin-AP Fab fragments antibody (1:1,500, 11093274910, Sigma-Aldrich). The next day, targeted mRNA-expressing cells were visualized as a dark purple deposition using the NBT/BCIP–alkaline phosphatase reaction (11681451001, Sigma-Aldrich).
Immunohistochemistry staining of human post-mortem brain tissue
To classify demyelination in post-mortem human brain tissue, the sections were stained using immunohistochemistry with diaminobenzidine (DAB, SK-4105, Vector Laboratories) for the detection of myelin oligodendrocyte glycoprotein (MOG). Tissue sections were initially fixed in 100% methanol at −80 °C for 5 min at room temperature, followed by a 5 min PBS wash at room temperature. A hydrophobic barrier was then created around the sections using a PAP pen (VEC-H-400, Biozol), after which they were blocked with 10% goat serum (16210064, Thermo Fisher Scientific) diluted in 0.01% PBS/Triton X-100 for 30 min at room temperature. After the blocking step, sections were incubated overnight at 4 °C with the primary MOG antibody (MAB5680, Merck Millipore). The next day, the sections were rinsed with PBS and then incubated for 2 h at room temperature with a biotinylated secondary IgG antibody. After 2 additional PBS washes, the sections were treated with an avidin-biotin complex for 1 h at room temperature. The DAB substrate was applied until a colour change was visible under the microscope. Subsequently, the sections were dehydrated using a gradient series (50%, 70%, 96%, 100%) of ethanol and cleared with two 10-min incubations in 100% xylene. Finally, the sections were mounted using Eukitt (Orsatec).
Human fresh frozen brains sections were fixed in 4% PFA for 10 min at room temperature. After fixation, the sections were rinsed three times with 1× PBS to remove any residual fixative. A hydrophobic barrier was created around the tissue sections using a PAP pen. The tissue sections were simultaneously blocked and permeabilized by incubating them in 5% goat serum diluted in 0.1% PBS-Triton X-100 for 1 h at room temperature. After blocking, the sections were briefly washed with 0.05% PBS/Tween-20 for 2 min. The primary antibodies (Supplementary Table 11) were incubated overnight at 4 °C in 0.05% PBS/Tween-20. The next day, the slides were washed three times with 1× PBS for 2 min each, followed by a secondary antibody (Supplementary Table 11) incubation for 1 h at room temperature. After secondary antibody staining, the slides were washed again with 1× PBS for 2 min. Finally, the sections were mounted using Fluoromount mounting medium containing DAPI (15596276, Invitrogen).
Immunocytochemistry
Cells were cultured in glass bottom 24-well plates (P24-1.5H-N, Cellvis), washed twice with 1× PBS and then fixed in 4% PFA for 15 min at room temperature. Fixed cells were washed in 1× PBS for 10 min thrice. Blocking solution (10% normal donkey serum in 0.1% Triton X-100/1× PBS) was added for 30 min at room temperature before primary antibodies (Supplementary Table 11) were added to blocking solution and incubated with fixed cells for 3 h at room temperature. Antibody solution was removed and the wells washed in 1× PBS for 10 min thrice. Secondary antibodies (Supplementary Table 11) were diluted in 0.1% Triton X-100/1× PBS and incubated for 1 h. Antibody solution was removed and the wells washed in 1× PBS for 10 min thrice. Wells were incubated with Hoechst (1:1,000, X) for 5 min at room temperature followed by three more washes in 1× PBS for 5 min each.
smFISH
Single-molecule fluorescence in situ hybridization (smFISH; RNAscope) analysis of rodent tissue sections was performed using the RNAscope LS Multiplex Assay Kit (Advanced Cell Diagnostics, ACD) as previously described2. z-probes with one of four distinct tail configurations (C1–C4) were obtained from ACD (Supplementary Table 12). Slides with cryosections were thawed at room temperature for 15 min before incubating at 65 °C for 45 min in an oven. Slides were immersed in 4% PFA at 4 °C for 15 min and then washed twice with 1× PBS. Slides were then transferred through an ethanol solution series: 5 min each at 50%, 70%, 100% and 100% ethanol in water. Coverplates (S21.4611, Leica) were added and slides were inserted into the Leica BOND RX. The slides were then exposed to ER2 (AR9640, Leica) at 95 °C for 5 min and subsequently, ACD Protease III at 42 °C for 20 min. ACD hydrogen peroxide was added for 10 min at room temperature to inactivate endogenous peroxidases and ACD protease III. Probe master mixes were produced by diluting z-probes in ACD Blank Probe Diluent master (300048, Biotechne). Probe mixes were hybridized to sections for 2 h at 42 °C. Sequential incubations in AMP1, AMP2 and AMP3 reagents for 30 min each at 42 °C allowed branched DNA tree development. The samples were then incubated with their corresponding HRP reagent for 1 min at 42 °C. TSA Opal 520, TSA Opal 570, TSA Opal 650 and TSA Opal 690 (Akoya Biosciences, FP1487001KT, FP1488001KT, FP1496001KT, FP1497001KT) were incubated at 1:500 concentration in TSA buffer for 30 min at 42 °C, followed by blocking with HRP blocking reagent for 15 min at 42 °C. For probes using ATTO 425 dye, the probe was incubated with TSA-biotin (Akoya, 1:500) for 30 min at room temperature, followed by streptavidin conjugated ATTO 425 (Sigma-Aldrich, 1:400) for 30 min. The samples were incubated in DAPI (0.25 μg ml−1, Sigma-Aldrich) for 20 min at room temperature. The slides were removed from the Leica BND RX and coverslipped with ProLong Gold Antifade Mountant (P36930, Thermo Fisher Scientific).
To identify human neurotypical CUX2 and ATF4 co-expression, the slide preparation protocol was modified as follows: the slides were first baked for 20 min at 60 °C before fixing with 4% PFA for 10 min. After a PBS wash of 5 min, slides were incubated in hydrogen peroxide and protease reagent (322381, ACD Bio) for 10 min, then subjected to antigen retrieval in Target retrieval buffer (322000, ACD Bio) at 95 °C for 5 min. The sections were then washed in distilled water and dehydrated in 100% ethanol. 3-plex RNAscope was performed using tyramide signal amplification with Opal 520, 570 and 690 dyes to label the probes.
Immunoblotting
Cells were collected in RIPA buffer (Sigma-Aldrich, R0278) supplemented with 1× protease inhibitor cocktail (Thermo Fisher Scientific, 78425), incubated on ice for 30 min with intermittent vortexing. Lysates were frozen at −80 °C until processing. The samples were centrifuged at 13,000g for 10 min at 4 °C and the supernatant was collected and suspended in 1× Bolt LDS sample buffer (Thermo Fisher Scientific, B0007) and 1× Bolt sample reducing agent (Thermo Fisher Scientific, B0009) before being boiled at 70 °C for 10 min. Proteins were separated by gel electrophoresis within 4–12% Bolt Bis-Tris gels (Thermo Fisher Scientific, NW04125BOX) before being transferred to PDVF membranes at a constant 12 V at 4 °C overnight. Membranes were blocked in 5% non-fat milk diluted in 1× Tris-buffered saline with 1% Tween-20 (TBST) for 1 h, washed twice and incubated with primary antibodies (1:1,000; Supplementary Table 11) diluted in 1× TBST overnight at 4 °C. After three 10 min washes in 1× TBST, membranes were incubated in HRP conjugated antibodies (Supplementary Table 11) diluted in 1× TBST for 1 h at room temperature. After three more washes in 1× TBST, membranes were reacted with SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Scientific, 34580) and visualized on a ChemiDoc system (Bio-Rad). Phosphorylated ATM bands were stripped using Restore Western Blot Stripping buffer (Thermo Fisher Scientific, 21059) before membranes were probed for total ATM. Bands were identified and quantified in ImageLab software (v.6.1, Bio-Rad).
Imaging
Standard imaging of immunohistochemistry or in situ hybridization was performed on the ZEISS Axio Imager 2 using Zen Blue Pro software (v.2.6). Confocal images were acquired on an Operetta CLS High Content Analysis System (Perkin Elmer) using the spinning-disc confocal mode with a sCMOS camera and a ×40/1.1 NA apochromatic water dispensing objective with eight separate LED light sources and acquired using Harmony software (v.4.9). Each z-stack consisted of exactly 21 planes with a 1 µm step size and tiled images were taken with 3% overlap. Images were stitched using a Perkin-Elmer tool (Acapella v.5.3.1).
For DNA damage immunohistochemistry and any immunocytochemistry, images were acquired on a Leica SP5 confocal system fitted with DMI8 inverted microscope stand and a 40×/1.3 NA Plan Apo objective using a 405 nm diode laser, 458, 488 and 514 nm lines of an argon laser, plus 561 nm HeNe and 633 nm HeNe lasers and detected with 2 PMT and 2 Leica HyD detectors. Images were stitched and projected using Leica Application Suite X (v3.5.7.23225) software. For imaging of post-mortem human RNAscope, a Leica Stellaris SP5 confocal system was used. Leica Stellaris confocal system was fitted with DMI8 inverted microscope with a HC PL APO CS2 ×40/1.30 NA oil objective, Diode 405, Diode 638, OPSL488 and OPSL 561 nm lasers and detected with Trans PMT and Leica HyD detectors. Images were stitched and projected on Leica LASX software. Stained sections from post-mortem human brain samples were visualized using a Leica DM6 B Thunder microscope equipped with a Leica K5C camera. Images were collected at ×40 magnification for high-resolution visualization of the target probes. To capture the entire signal within the tissue sections, images were acquired as z-stacks based on Nyquist sampling criteria.
Layer thickness and cortical cell counts
Regions of interest were analysed from 1–3 consecutive 16 μm sections over the primary somatosensory barrel field and averaged. Layers were identified by cell morphology changes based on both Hoechst or DAPI in combination with NeuN staining. Layer thickness was measured in Fiji v.2.14.0/1.54f (ImageJ.net) and cell counts were manually taken using the ImageJ Cell Counter plugin and averaged. The researcher was blinded to the experimental conditions.
Quantification of DNA damage marks
Regions of interest from the mouse cortical L2/3 (1.2 × 105 μm from 1–3 consecutive 16 μm sections) and the human cortical L2/3 (4.9 × 105 μm) were analysed for 53BP1+ and γH2AX+ foci within NeuN stained cells. The ImageJ Cell Counter plugin (on Fiji v.2.14.0/1.54 f) was used to track cell characteristics and the number of γH2AX+ foci was manually recorded per cell. The researcher was blinded to the experimental conditions, with the exception that two human neurotypical controls were added after the initial blinded analysis because one of the blinded neurotypical samples had to be removed as the brain showed Alzheimer’s pathology and cortical atrophy.
Study design and statistical analysis
Sample size was determined based on the likelihood of phenotypic effects observed in previous studies from the same human samples and animal models used2,17,18,20,52,53,54,55,58,71, while also considering the availability of sex and age matched samples. All sample sizes are individually reported in figure legends. All data were produced from independent repeated experiments using biological replicates as stated in the figure legends and methods. All samples were randomized with the exception that equal male and female mice were allocated to experimental groups amongst littermates. Experimenters were not fully blinded to group allocation during data collection, but sample IDs without group information were used to deter bias and all experimental parameters were kept consistent across groups, so there are no differences between replicates/groups. Experimenters were blinded to the experimental parameters during data analyses unless the analyses involved automated quantification whereby experimental parameters were applied consistently across all samples. Two human neurotypical controls were analysed after the initial blinded analysis as one blinded sample was removed due to conflicting Alzheimer’s pathology. All seven remaining samples were quantified with the researcher blinded to the experimental condition. Subjective measurements were not used in this study.
Statistical tests on RNA-seq data were determined using Wilcoxon rank-sum tests performed in R software unless otherwise stated in the figure legends. Statistical analysis on cell culture and histological data were performed in Prism software (v.10.4.1). The samples were first tested for normality and then statistical comparisons were made using either the parametric or nonparametric counterparts as reported in the figure legends. Images were pseudocoloured in Adobe Photoshop (v.26.4.0) and the brightness and contrast were adjusted consistently across samples. The figures were finalized in Adobe Illustrator (v.30.1).
Materials availability
All unique materials generated in this study can be obtained from the lead contact upon completion of a materials transfer agreement.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
First Appeared on
Source link

