A repeat expansion in GOLGA8A is a major risk factor for atypical frontotemporal lobar degeneration with ubiquitin-positive inclusions
FTLD-FET consortium
We established an international consortium to identify and bring together a sufficiently large case population to systematically assess this group of rare disorders. FTLD-FET patients were identified through inquiries at brain banks focused on neurodegenerative disease research and by contacting authors of relevant publications. All patients or their next of kin provided consent to participate in research studies in accordance with the Declaration of Helsinki and local ethics review board standards at each of the participating sites. The ethics committee of the University Hospital Antwerp and the University Antwerp approved the study. Our primary goal was to identify aFTLD-U cases; however, small numbers of NIFID (n = 33) and BIBD (n = 12) cases were identified and collected during these efforts (Supplementary Table 4).
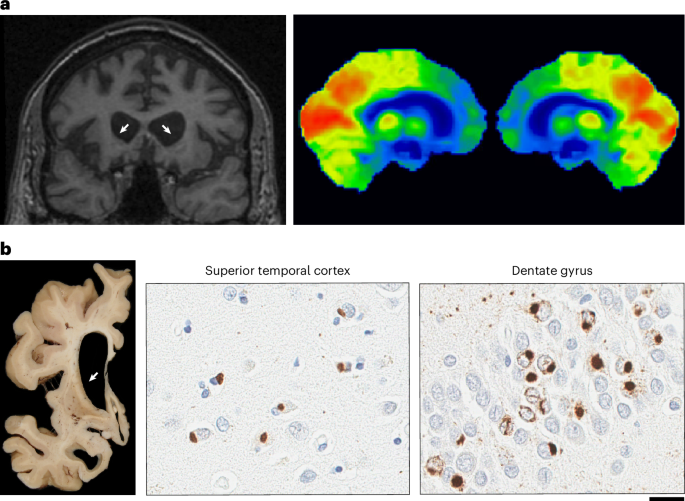
An experienced neuropathologist from one of the collaborating sites analyzed paraffin-embedded tissue sections for each patient to confirm the neuropathological diagnosis. As our genetic studies primarily focused on aFTLD-U, the patient characterizations were focused on differentiating aFTLD-U from the other FTLD-FET diagnoses. Specifically, aFTLD-U was diagnosed based on the presence of tau- and TDP-43-negative, FUS-positive neuronal cytoplasmic inclusions (NCI) and FUS-positive neuronal intranuclear inclusions (NII). FUS immunostaining was performed at most sites using primary antibodies 11570-1-AP (Proteintech Group) and/or HPA008784 (Sigma Life Sciences), and occasionally A300-302A (Bethyl Laboratories) or aa1-50 (Novus). None of the aFTLD-U cases showed basophilic inclusions (characteristic of BIBD) or other cellular inclusions, such as hyaline conglomerate inclusions (typical of NIFID), on hematoxylin and eosin staining. The diagnosis of aFTLD-U was further supported by the presence of only limited FUS pathology in subcortical regions and limited variability in the morphology of NCIs. In those cases where a differential diagnosis of NIFID was considered, neurofilament or alpha-internexin (AIN) immunohistochemistry was performed to exclude a pathological diagnosis of NIFID. In a minority of aFTLD-U cases, TAF15 immunohistochemistry (A300-308, Bethyl Laboratories) was also performed, confirming TAF15 immunoreactivity (of the inclusions).
So far, our collective efforts have identified 108 aFTLD-U cases from 24 sites, and new cases are being added regularly. The mean age at onset in the full cohort was 44.3 years (median 43, standard deviation 10.4 years, range 21–73 years), with the mean age at death of 51.0 (median 51, standard deviation 10.0 years, range 30–77 years) and a mean disease duration of 6.8 years (median 6, standard deviation 3.4, range 2–19 years). We observed a notable sex imbalance of 34 (31.5%) female and 74 (68.5%) male aFTLD-U cases, which was not observed in NIFID (female n = 16, 48.5%; male n = 17, 51.5%) or BIBD cases (female n = 7, 58%; male n = 5, 42%). All cases were self-reported Caucasian except for one aFTLD-U case of Asian ancestry.
Frozen brain tissue from the cerebellum and/or frontal cortex was obtained from 84 aFTLD-U cases, while DNA extracted from blood was available from four additional aFTLD-U cases. For a small subset of aFTLD-U cases, multiple brain regions and LCLs generated by Epstein-Barr virus transformation were available. Only fixed tissue was available for the remaining 20 aFTLD-U cases. A source of DNA was available from 23 out of 33 NIFID cases and 11 out of 12 BIBD cases (Supplementary Table 4).
Inquiry at participating sites also identified a source of DNA (blood or LCL) from five relatives (four siblings and one child) related to three different aFTLD-U cases.
Additional cohorts
To establish population frequencies of the disease-associated haplotypes A and B and characterize their repeat lengths and sequence composition in non-FTLD-FET and control cohorts, we used several additional populations summarized in Supplementary Tables 1 and 4. These included an in-house cohort of FTLD-TDP cases and controls previously included in long-read sequencing projects, a Mayo Clinic control population including both neuropathologically confirmed normal individuals as well as a clinical cohort of neurologically healthy controls, a cohort of patients with other neurodegenerative diseases (progressive supranuclear palsy, Lewy body dementia and multiple system atrophy) from the Mayo Clinic brain bank (Mayo non-aFTLD-U), Alzheimer’s disease cases and controls from the European Alzheimer’s Disease DNA BioBank (EADB), and individuals from the Oxford Nanopore Technologies (ONT) 1000 Genomes Project19,20,22. From the cohort of the ONT 1000 Genomes Project, we identified one repeat expansion carrier who passed on haplotype A to his daughter, for whom only short-read sequencing data was available. We requested an LCL sample from the daughter from the Coriell biobank for long-read sequencing.
The Belgian EADB cohort includes Alzheimer’s disease cases ascertained at the Memory and Neurology Clinics of the BELNEU consortium, and cognitively healthy control individuals who were partners of patients or volunteers from the Belgian community23. All control individuals scored >25 on the Montreal Cognitive Assessment test and were negative for subjective memory complaints, neurological or psychiatric antecedents, and family history of neurodegeneration. All participants and/or their legal guardian signed written informed consent forms before inclusion. The study protocols were approved by the ethics committees of the Antwerp University Hospital and the University of Antwerp, and the ethics committees of the participating neurological centers of the BELNEU consortium. Genotyping was performed using the Illumina Infinium Global Screening Array (GSA, GSAsharedCUSTOM_24 + v1.0). Details on quality control, variant calling and imputation have been described in detail by Bellenguez et al.18.
Short-read genome sequencing
DNA samples from 23 aFTLD-U cases and 1,304 neurologically normal controls were sequenced using short-read genome sequencing (phase I) as part of efforts related to the International FTLD-TDP whole-genome sequencing consortium8,26. In brief, DNA from 982 control participants from the Mayo Clinic Biobank were sequenced at HudsonAlpha using the standard library preparation protocol using NEBNext DNA Library Prep Master Mix Set for Illumina (New England BioLabs) on Illumina’s HiSeq X. Before analysis, participants from this cohort with possible clinical diagnosis or family history of a neurodegenerative disorder were removed (n = 144 removed; n = 838 remaining). Whole-genome sequencing for the 23 aFTLD-U cases was performed at the USUHS Sequencing Center, and 322 controls free of neurodegenerative disorders were sequenced at Mayo Clinic Rochester using the TruSeq DNA PCR-Free Library Preparation Kit (Illumina), followed by sequencing on Illumina’s HiSeq X. In a next phase, genome sequencing of 38 newly ascertained aFTLD-U cases (phase II) was performed at Mayo Clinic Rochester using the Nextera DNA Flex Library prep kit followed by sequencing on Illumina NovaSeq. To enhance our study, we further incorporated genomic variant call format (gVCF) files from 2,037 control individuals obtained from the Alzheimer’s Disease Sequencing Project (ADSP). gVCF enables joint genotyping with the existing cohort, as those files provide a comprehensive record of variant calls and reference positions. The gVCF files from ADSP controls were merged with our cohort’s gVCF files using the joint-genotyping approach implemented with the Genome Analysis Toolkit (GATK). By merging these gVCFs, we ensured all our patients and controls were analyzed together, allowing a more robust comparison and reducing batch effects.
For all cases and all controls except those from ADSP, fastq files were processed through the Mayo Genome GPS v4.0 pipeline. Reads were mapped to the human reference sequence (GRCh38 build) using the Burrows-Wheeler Aligner34, and local realignment around indels was performed using the GATK. Variant calling was performed using GATK HaplotypeCaller followed by variant recalibration (VQSR) according to the GATK best practices35. Variant calling on the final dataset for analysis included the gVCF from 2,037 ADSP control individuals to allow joint genotyping of all cases and controls.
No pathogenic variants in genes linked with neurodegenerative disorders were identified in the aFTLD-U cohort based on genome sequencing and repeat-primed PCR for the C9orf72 repeat expansion36. Mutations in the coding exons of FUS and TAF15 were excluded by Sanger sequencing in patients for whom no genome sequencing data were generated.
Sample-level quality control
Samples with less than 30× coverage in more than 50% of the genome, call rate below 85%, sex error, or contamination defined by a FREEMIX score above 4 were removed. After joint genotyping of all samples, relatedness was assessed using KING37, duplicates were removed and only one individual per family (second-degree relatives or closer) was kept. Individuals with <70% European ancestry based on Admixture analysis were removed38. In the aFTLD-U cohort, one case had too low coverage and one Asian case failed ancestry quality control. In total, 59 aFTLD-U cases and 3,153 control individuals passing all quality control measures were included in the analysis (Fig. 2d).
Variant-level quality control
Genotype calls with genotype quality <20 and/or depth <10 were set to missing, and variants with overall call rate <80% were removed. Gene annotation of variants was performed using ANNOVAR (version2016Feb01).
Generation of principal components
Before running genetic association analyses, principal component (PC) analysis was performed using a subset of variants meeting the following criteria: minor allele frequency >5% and full-sample HWE P > 1 × 10−5. Influential regions such as the HLA region were removed, and variants were pruned by linkage disequilibrium with an r2 threshold of 0.1. We generated PCs, and the top four PCs were included as covariates.
Genome-wide association analyses
GWAS was performed using REGENIE7, including SNVs with minor allele frequency >0.01 in cases or controls and HWE P > 1.0 × 10−6 in controls. Only variants that passed VQSR filter and with a call rate >90% in both cases and controls were included in the analyses. To remove spurious associations due to potential sequencing batch effects, further filters were applied. Batch effect tests were performed separately for controls (analysis of variance, P < 0.01) and cases (Fisher exact test, due to smaller groups), comparing genotype distributions and removing any variant with genotype frequency differences between batches in either cases or controls (P < 0.01).
For all remaining 6.9 M variants, the association of genotypes with the case/control status was assessed using REGENIE with allele dosage as the predictor assuming log-additive allele effects. Sex and the first four PCs were included as covariates in the models. We additionally performed a conditional GWAS analysis after removing carriers of the rs549846383 rare allele, applying the same filters described above but without filtering for HWE, testing for association in 7.4 M variants. Variants at chr15q14 were visualized with locuszoom39.
A separate cluster of control individuals was identified in the PC plot (Supplementary Fig. 2), and as a sensitivity analysis, we repeated the GWAS while removing those outlier controls, defined as all individuals that are three standard deviations removed on either PC1 and PC2 from the PC center.
Sanger sequencing genotyping and validations
The rs549846383 and rs148687709 haplotype tagging variants were genotyped using PCR and Sanger sequencing, with primer sequences in Supplementary Table 5. The assay for rs549846383 uses Titanium Taq (Takara Bio), 1 M betaine and 3 min at 95 °C, 32 cycles of 30 s at 95 °C, 30 s at 62 °C and 1 min at 68 °C, with finally 5 min at 68 °C in a Veriti 96-well fast thermal cycler (Applied Biosystems). The assay for rs148687709 is identical, except for a final concentration of 2 M betaine. The results of rs148687709 must be interpreted as tetraploid, as no unique primers could be designed, and the paralogous sequence in GOLGA8B will also be amplified (Supplementary Fig. 17). Sanger sequencing results were analyzed using Seqman (DNASTAR) and novoSNP40.
Long-read genome sequencing
Long-read genome sequencing on the PromethION P24 (ONT) was performed for 53 aFTLD-U cases and 5 non-aFTLD-U individuals carrying haplotype A selected from FTLD-TDP short-read genome sequencing and Mayo Clinic controls. For 49 cases, DNA was extracted from the frontal cortex, while DNA from the remaining cases was extracted from the cerebellum. The newly generated dataset was combined with an ongoing genome sequencing initiative of 283 non-aFTLD-U individuals, mostly FTLD-TDP patients and neurologically normal controls. In a second phase, 11 non-aFTLD-U individuals were sequenced, including 8 carrying haplotype A (4 patients with progressive supranuclear palsy, 1 patient with Lewy body dementia, 1 patient with multiple system atrophy and 2 neurologically healthy controls) and 3 neurologically healthy controls carrying haplotype B. An overview of the long-read sequencing cohorts can be found in Supplementary Table 1. We additionally sequenced the genome of one NIFID patient, and sequenced other brain regions (caudate, cerebellum and occipital cortex) and LCLs for selected aFTLD-U cases, as well as LCL- and blood-derived DNA from two unaffected siblings of two aFTLD-U cases. Finally, we requested an LCL sample from HG01514 from the Coriell biobank/NINDS Repository for long-read sequencing.
DNA was extracted from brain tissue using the Nanobind tissue kit (PacBio) and from LCLs with the Qiagen DNA Mini Kit, followed by quality control using the Dropsense (Trinean), Qubit (Thermo Fisher Scientific) and Fragment Analyzer (Agilent) to assess purity, concentration and fragment length. DNA was sheared using the Megaruptor 3 (Hologic, Diagenode) on speed 28–30, followed by removing short fragments with the Short Read Eliminator (PacBio) when considered appropriate. The library prep was generated using the SQK-LSK110 or SQK-LSK114 kit (ONT) according to the manufacturer’s instructions, except for longer incubation times for enzymatic steps, before sequencing on an R9.4.1 or R10.4.1 flow cell for 72 h.
The sequencing data was base called with guppy (for R9 flowcells, v6.7.3) or dorado (for R10 flowcells, v7.1.4, v7.2.13, v7.3.11 and v7.4.13) using the high-accuracy (HAc) base calling model (ONT), including cytosine methylation and hydroxymethylation inference. The data were processed using a snakemake workflow41 (github.com/wdecoster/chr15q14). Reads were aligned to the GRCh38 reference genome (GCA_000001405.15_GRCh38_no_alt_analysis_set) with minimap2 (v2.24)42, followed by sorting reads by coordinate and conversion to CRAM format with samtools (v1.16.1)43. The data quality was assessed with cramino (v0.14.5), as was the concordance with the expected sex based on the normalized read depth of the sex chromosomes44. Reads were phased with longshot (v0.4.5)45. SVs were called using Sniffles2 (v2.5.3)46 and SNVs with Clair3 (v1.0.2)47 and Deepvariant48, followed by merging variants in gvcf format using GLnexus49 and annotation using VEP50.
We performed ultralong nanopore sequencing for two participants, a sib pair sharing the haplotype with one affected and one unaffected individual (Fig. 5a, FAM1). DNA was extracted from LCL pellets, following the SQK-ULK114 protocol (ONT) with sequencing on the PromethION and super accuracy base calling (dorado v7.3.11). Obtained data were combined with the standard long-read genome sequencing data (SQK-LSK114), filtered for reads longer than 25 kb using chopper (v0.8.0)44 and assembled with hifiasm (v0.24.0-r703) with the –ont option51, followed by SV calling with svim-asm (v1.0.3)52.
Tandem repeat analysis
Tandem repeats of interest were genotyped with STRdust (v0.11.7)53, either from local files as sequenced in-house or over FTP for the participants from the 1000 Genomes Project resequenced with ONT19,20,22. STRdust was used in standard (phased) mode to establish that the repeat expansion is present on the associated haplotype. As read phasing by LongShot was found to be unreliable for this locus, resulting in the omission of a large proportion of the reads from the phased results due to ambiguous alignment and uncertain haplotype assignment, the unphased mode of STRdust was used to obtain the genotypes used in this Article, determining alleles by hierarchical clustering the extracted repeat sequence for each read. STRdust generates a consensus allele by partial overlap alignment as implemented in rust-bio54, ignoring length outliers. The observed length variation suggests that the consensus sequence can change substantially due to random sampling of sequenced fragments from the library, especially at low sequencing depth.
The length of all human tandem repeats55 was determined using inquiSTR (v0.13.0) (github.com/wdecoster/inquiSTR). We developed STR_regression.R (v1.6) (github.com/wdecoster/inquiSTR/scripts/STR_regression.R) for running association testing of tandem repeat lengths, which can fit generalized linear models using the output of inquiSTR repeat lengths and phenotypic information of multiple samples. STR_regression.R can run both logistic and linear regressions based on binary and continuous phenotypes (and optionally with covariates), and it outputs detailed statistics of repeat length associations. Moreover, it has multiple functionalities, including different repeat length processing modes (considering either mean, minimum or maximum repeat length for a given tandem repeat), various run options (genome-wide, per chromosome and a region of interest based on a chromosomal interval or a list of regions of interest based on a BED file), and it can also take into account provided cutoffs to define expanded alleles of tandem repeats. For this analysis, we compared 52 aFTLD-U cases with 283 non-aFTLD-U individuals, excluding one Asian aFTLD-U case and the five haplotype-A-carrying non-aFTLD-U individuals specifically selected for long-read sequencing. We used the longest allele per individual for all human tandem repeats, with a binary phenotype (aFTLD-U or not), a minimal call rate of 80% and Bonferroni correction for multiple testing.
The repeat composition was assessed using a k-mer heatmap, in which all 12-mers were quantified. As the CCCCT pentamer expansion was found in only a single case, the repeat composition in the cohort was quantified and visualized using the least common multiple of 12-mer units to simultaneously represent dimer, tetramer and hexamer motifs, that is, the most commonly observed motifs. VCF files were parsed with cyvcf2 (v0.30.16)56, and each 12-mer in the repeat consensus sequences was counted. After counting, all motifs were rotated and represented by the lexicographical first, then collected in a pandas dataframe57 before filtering motifs rarely observed, except if highly prevalent in one individual. Visualization was done using Plotly (v5.14.1)58. We also used aSTRonaut (v1.0)53 to visualize the sequence of the observed repeat motifs per allele (CT, CCTT, CTTT, CCCT, CCCTCT, CCCCT, CCTTT and CCCCCC), replacing motifs by colored dots of the same length, substituting longer motifs first.
We calculated the CT dimer count for each repeat allele by removing all occurrences of other repeat motifs in which CT is a substring (CCCTCT, CCCCT, CCTT, CCCT and CTTT) from the consensus allele and counting the remaining CT units. Precision and recall of the proposed cutoffs (>190 CT dimers or >450 bp repeat and >80% CT) was calculated using scikit-learn (v1.6.1)59 with CIs calculated using bootstrapping as implemented in scipy (v1.15.1)60.
Copy number variant analysis
The copy number of the region between GOLGA8A and GOLGA8B (chr15:34438297–34524132), which is a unique sequence in the human reference genome, was quantified using the coverage obtained from mosdepth (v0.3.8)61, normalized to a copy-number-neutral interval (chr15:54033377–56279876) for both short- and long-read genome sequencing data. Visualization was performed in Python using Plotly (v5.14.1)58, and statistical analysis was performed for carriers of the deletion allele using a Fisher exact test as implemented in scipy (v1.15.1), comparing the deletion versus normal copy number for aFTLD-U cases against controls60.
Phylogenetic analysis
A phylogenetic tree of haplotypes in the locus of interest (defined as 500 kb surrounding the main tagging variant, chr15:34362469–34862469) was generated using the process described below. First, variants were called with Deepvariant48 (v1.8.0) and phased with whatshap62 (v2.8). We then selected samples that were fully phased in one phaseblock for the locus of interest using phasius44, and removed samples with a copy number suggestive of a deletion or a duplication (removing samples with a normalized copy number below 0.8 or above 1.2). Subsequently, reads were tagged with the haplotype identifier (whatshap haplotag), then splitting the bam file into two haplotypes with samtools split43 (v1.13). A consensus in fasta format was generated for each haplotype using samtools consensus, for which then a multisequence alignment was generated using mafft63 (v7.526), followed by generating a phylogenetic tree with iqtree64 (v2.4.0). The obtained tree was then visualized using ggtree65 (v3.14.0).
Southern blotting
The length of the repeat expansion was confirmed with Southern blotting, using a 437-bp PCR probe, generated from genomic DNA using the PCR DIG Probe Synthesis Kit (Roche) and the following primers: forward: GGACCCTTTAGAGTTGCTTC and reverse: GTATGGAGGGCAGAGTTGTTG (corresponding to chr15:34,420,657–34,421,094). With this configuration, the expected (reference) DNA fragment size is ~4.2 kb. Genomic DNA was extracted from frontal cortex tissue, and 8 μg was digested overnight with Kpn1 and electrophoresed in a 0.8% agarose gel for 6:30 h at 100 V. The DNA was transferred to a positively charged nylon membrane (Roche) by 20-h capillary blotting and then crosslinked by ultraviolet irradiation. Prehybridization in 20 ml DIG EasyHyb solution for 3 h was followed by overnight hybridization at 47.8 °C in a shaking water bath with 30 μl of PCR-labeled probe in 7 ml of DIG EasyHyb. The membrane was washed twice in 2× standard sodium citrate, 0.1% sodium dodecyl sulfate at room temperature for 5 min each, and twice in 0.1× standard sodium citrate, 0.1% sodium dodecyl sulfate at 68 °C for 15 min each. Detection of the hybridized probe DNA was done as described in the DIG System User’s Guide (Roche). CDP-star chemiluminescent substrate was used, and signals were visualized on X-ray film after 30–60 min. The ladders used are the DNA Molecular Weight Marker II with fragments at 23,130, 9,416, 6,557, 4,361, 2,322, 2,027, 564 and 125 bp, and the DNA Molecular Weight Marker VII with fragments at 8,576, 7,427, 6,106, 4,899, 3,639, 2,799, 1,953 and 1,882 bp, and nine smaller bands.
Repeat-primed PCR
The genomic region on chr15q14 containing the expanded alleles was amplified using a panel of three-primer repeat-primed PCR assays, each with one FAM-labeled primer flanking the repeat, one sequence-specific primer targeting each of the repeat motifs and one booster primer recognizing the tail of the sequence-specific primer to amplify the signal. A total of six primer sets were designed based on observed repeat sequences (Supplementary Table 5), in particular, to determine the presence of CT motifs on the left and right ends of the repeat, CCCTCT motifs on the left and right ends, CCCT motifs on the left, and CCCCT motifs on the left end of the repeat.
The primers are used in equal proportions with amplification using the PrimeSTAR GXL DNA polymerase kit (Takara). Initial denaturation was performed for 2 min at 98 °C, followed by 36 cycles of 10 s at 98 °C, 15 s at 58 °C, and 1 min at 68 °C, with a final extension of 3 min at 68 °C. Fragment lengths were determined with capillary electrophoresis on an ABI3730XL using an internal size standard (LIZ500HD, Thermo Fisher Scientific) and visualized using the in-house developed traci software (v1.1.0) (https://github.com/derijkp/traci).
Ethics and inclusion statement
As FTLD-FET is a rare disorder, this study was made possible only through a large international collaboration. All colleagues from local sites fulfilling authorship criteria are included in the author list.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
First Appeared on
Source link

