Ectopic expression of cytosolic DHODH uncouples de novo pyrimidine biosynthesis from mitochondrial electron transport
Cell culture
Cells were grown in complete DMEM medium (D5796, containing 4,500 mg l−1 glucose, 2 mM L-glutamine) supplemented with dialysed 10% FBS (Sigma-Aldrich, F7524), 1% penicillin–streptomycin (PenStrep, Lonza), at 37 °C in an atmosphere of 5% CO2 and 95% air. Where indicated, 50 μg ml−1 uridine and/or 1 mM sodium pyruvate (Sigma-Aldrich) were added to the growing media. Inhibitors were purchased from Sigma-Aldrich and used as follows: brequinar (2 μM); antimycin A and myxothiazol (20 μM each); and chloramphenicol (40 μg ml−1). Cells were pre-treated with chloramphenicol for 1 week in the presence of uridine to deplete them of respiratory complexes48. Control cells were treated with the appropriate vehicle (ethanol or dimethylsulfoxide (DMSO)). The origin of cell lines was: ρ0 cells49, Mt-Cybmut and Cox10KO AOX-expressing cells29.
Generation of ScURA-expressing cells
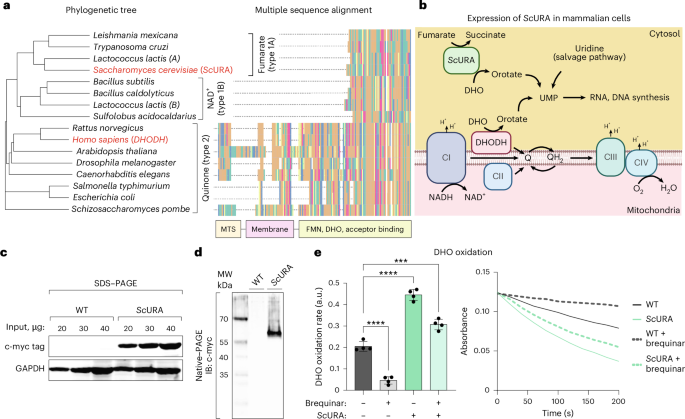
The coding sequence of ScURA was obtained from a previous publication50 and modified as follows. A myc tag was added at the C terminus through a (GGS)3 linker, and at both ends, restriction enzyme sites were inserted. Codon-optimization and oligonucleotide synthesis were performed by Genewiz. The gene was cloned into the pWXLd lentiviral vector (modified from Trono’s lab with a puromycin resistance gene, Addgene). Lentivirus production was carried out by the Viral Vector unit at Centro Nacional de Investigaciones Cardiovasculares (CNIC). Viral supernatants were filtered and added to cells with 8 μg ml−1 polybrene overnight. Then, 2 days after transduction, ScURA–expressing cells were selected with puromycin 0.5 μg ml−1 for approximately 1 week until non-transduced cells in a control plate were dead. For each cell line, a cell line transduced with an empty vector was used as a control.
Growth curve and cell doubling time
On day 0, a total of 40,000 cells were plated into six-well plates. For each condition, three wells were incubated for 6 h (to allow the cells adhere to the plate), collected and counted. This count was recorded as the number of cells at day 0. For 3–4 days, fresh media and the appropriate treatments were added to each well every day. On the last day, three wells per condition were collected and counted. The number of doublings pre day was calculated as 24 / ((T × (ln2)) / (ln(nf / n0))), where T is time in hours, nf is the number of cells on the final day and n0 is the number of cells at day 0.
Whole-cell homogenate and mitochondria isolation
Cells from two to ten 150 mm plates were collected with trypsin, washed in PBS and resuspended in cold sucrose buffer (0.32 M sucrose, 1 mM EDTA, 10 mM Tris-HCl pH 7.4). Cells were then homogenized in a Teflon potter-type tissue homogenizer with 20–40 ‘pops’. The homogenate was centrifuged at 1,000g for 5 min, the nuclear pellet was discarded and the post-nuclear whole-cell homogenate was stored at −80 °C. When mitochondria were isolated, the whole-cell homogenate obtained was further centrifuged at 10,000g for 10 min. The pellet obtained was resuspended in sucrose buffer and stored at −80 °C.
Spectrophotometric DHODH activity
DHODH activity was measured following a previously published method51, with some modifications: 100 µg of whole-cell homogenates were used for each reaction in a final volume of 200 µl. Each biological replicate was split into three technical replicates. DHODH-dependent reduction of 2,6-dichloroindophenol (DCPIP) was measured with a UV-visible spectrophotometer equipped with a 96-well plate reader. Samples were resuspended in reaction buffer (50 mM Tris-HCl, 150 mM KCl, 3 mM KCN, 0.05 mM DCPIP, 0.25 mM decylubiquinone, pH 7.5) and incubated for 2 min at 37 °C. The reaction was started by the addition of 1 mM DHO. The first 3 min of the reaction were linear and were used for the analysis. The rate of background (DHODH-independent) DCPIP reduction was measured in parallel by omitting DHO from the reaction mixture and was subtracted from the DHO-dependent rate for each sample. Brequinar was added where indicated at 0.05 mM; controls were treated with the same volume of DMSO.
Gel electrophoresis
All gels were prepared in-house with Tris-HCl buffer and acrylamide/bis. Protein samples were quantified and loaded on the gel, and electrophoresis was performed in Tris-glycine solution. For denaturing electrophoresis, 0.1% SDS was added to the gels and protein samples were incubated for 1 min at 95 °C in loading buffer (50 mM Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, 1% β-mercaptoethanol, 0.02% bromophenol blue) before loading. For non-denaturing native gels, SDS was not added to the gel, whole-cell homogenates were not boiled and the loading buffer used did not contain SDS or β-mercaptoethanol.
Immunoblotting
Proteins were transferred to PVDF membrane (Immobilon-FL, 0.45 μm) by transfer in BioRad Mini Trans-Blot Cell or Trans-Blot Cell systems, in 48 mM Tris, 39 mM glycine and 20% methanol transfer solution overnight at 30 V. The membrane was then blocked in 0.1% PBS-Tween, 5% BSA solution for 1 h at room temperature (22 °C) and incubated with primary antibody overnight at 4 °C. After that, the membrane was incubated with secondary antibody for 1 h at room temperature. Membrane development was performed using fluorescent secondary antibody and revealed with the Odyssey imaging system (LI-COR Biosciences). Antibodies used included anti-hemoagglutinin-tag (1:5,000; Sigma-Aldrich, sAB 4300603), anti-betaActin (1:10,000; Sigma-Aldrich, A2066), anti-GAPDH (1:5,000; Abcam, AB8245) and anti-SDHA (1:5,000; Invitrogen, 459200 or Proteintech, 14865-1-AP).
Blue native–PAGE and complex I in-gel activity
The procedure was carried out as detailed previously48. In brief, 100 μg of mitochondria were incubated with 400 μg of digitonin for 5 min in 50 mM NaCl buffer, 50 mM imidazole, 5 mM aminocaproic acid at a final concentration of 10 μg μl−1. Electrophoresis was performed in a cold chamber and developed for 30 min at 90 V with cathode buffer A. Then, the cathode buffer was exchanged for cathode buffer B, and electrophoresis continued for approximately 1 h more at 300 V. Measurement of NADH dehydrogenase activity of complex I was determined on the same gel after blue native–PAGE electrophoresis by incubating the gel in 0.1 M Tris-HCl, pH 7.4, 0.14 mM NADH and 1 mg ml−1 NitroBlue tetrazolium solution at room temperature.
ATP synthesis
ATP synthesis was assessed as previously described52. In brief, 1.5 billion cells were permeabilized with digitonin and ATP synthesis was assayed using glutamate plus malate or succinate as fuels in the presence of adenosine 5′-diphosphate and diadenosine pentaphosphate (to inhibit the adenylyl kinase) by a kinetic luminescence assay using the luciferin–luciferase reaction.
Oxygen consumption rate
A total of 10,000 cells were plated in a Seahorse XF96 microplate the day before the assay. Cells were washed in MAS medium (70 mM sucrose, 220 mM mannitol, 5 mM KH2PO4, 5 mM MgCl2, 1 mM EGTA, 2 mM HEPES pH 7.4) containing 0.2% w/v free fatty acid BSA and then placed in MAS media containing ADP and digitonin (4 mM and 10 µg ml−1 final concentrations, respectively), incubated for 3 min at 37 °C and then loaded in the instrument. Substrate injection was as follows: glutamate + malate (5 mM each, port A); rotenone (2 µM, port B); succinate (5 mM, port C); antimycin A (1 µM, port D). These conditions allow for the determination of the respiratory capacity of mitochondria through complex I and complex II. For measuring activities of mitochondrial complexes, frozen–thawed cells were used as previously detailed53.
Phylogenetic tree construction
The amino acid sequence of DHODH from various species was retrieved from UniProt. The R packages msa54, ape55 and ggmsa56 were used for multiple sequence alignment, phylogenetic tree construction and generation.
Immunocytochemistry
In total, 20,000 cells were seeded per well on a 12-well plate containing previously sterilized precision cover glasses (Marienfeld, 0117520) and left incubating at 37 °C and 5% CO2 in their corresponding medium. After 48 h, cells were fixed for 15 min at 37 °C in pre-warmed 4% paraformaldehyde (ThermoFisher, 043368.9M) diluted in 1× PBS, followed by three washes in 1× PBS. Fixed cells were permeabilized with 0.1% Triton-X100 in 1× PBS for 10 min at room temperature. Cells were blocked in 2% BSA (w/v) in 1× PBS, then incubated with a Tomm20-CL488 antibody (1:500) (Proteintech, 30235116) diluted in blocking buffer. Cells were then incubated in Alexa Fluor 405 N-hydroxysuccinimide (NHS) ester (1:5,000) (ThermoFisher, A30000) diluted in 1× PBS. All incubation steps were performed for 40 min at room temperature and followed by three 5 min washes in 1× PBS-Tween 0.1%. Samples were finally mounted on microscopy slides using ProLong Gold Antifade Reagent (Invitrogen, P36934).
Microscopy image acquisition
All microscopy images were collected on a Leica SP5 inverted confocal microscope equipped with a HCX PL APO λ blue ×63, 1.40 NA oil objective. Z-stacks containing 10 planes (with a step size of 0.34 µm) were acquired in a sequential line scanning mode using LAS-AF (v.2.6.0) software (Leica Microsystems) at a digital zoom of ×2, pixel size of 60 nm, scanner speed of 200 Hz and 3 line averages, with a pinhole size of 1 AU. The Tomm20 signal was excited using the Argon 488 laser line at 10% intensity, and the corresponding 505–555 nm emission was collected using a PMT detector set at 800 V gain and −0.1% offset. The NHS405 signal was excited using the UV diode 405 laser line at 2% intensity, and 420–485 nm emission was collected using a PMT detector at 900 V gain and −1% offset.
Microscopy image analysis
Image analysis was mainly performed in FIJI 1.53t. Z-stacks were first projected into a single plane using the maximum intensity method. For mitochondria, the signal of Ch2 (NHS405) was subtracted to that of Ch1 (Tomm20-CL488) and the resulting image was then thresholded with the Mitochondria Analyzer plugin57 using the following parameters: remove particles (size, ≤0.1 µm2), subtract background (rolling, 2.0), sigma filter plus (radius, 2.0), enhance local contrast (max slope, 3.0), weighted mean threshold (block size, 1.45; C-value, 5.0) and despeckle and remove outliers (pixels, 4.0). Individual networks were isolated from whole thresholded images, and morphology parameters of mitochondria were calculated on a per-cell basis. For analysis of cell morphology, the NHS405 channel was first pre-processed in FIJI by applying CLAHE (block, 500; bin, 256; slope, 10) and a Gaussian blur (radius, 0.3 µm). The objects were then thresholded and their morphology analysed in Cellprofiler (v.4.2.8)58 using an adaptive Otsu method with three classes.
CRISPR–CAS9-mediated generation of SDHAKO cell lines
U2OS (ATCC, HTB-96) and 143B (ATCC, CRL-8303) SDHAKO clonal cell lines were generated through nucleofection (Neon transfection system, Invitrogen) of ribonucleoprotein complexes prepared by incubating 2 µg of TrueCut Cas9 Protein v2 (Invitrogen, A36499) and 2 µg of a pair of single guide RNAs (sgRNAs) (sgRNA 1 target sequence at intron 1, AACCCTGAAGGCAGCCCAAG; sgRNA 2 target sequence at exon 4, GGGCCTGCTCCGTCATGTAG). DNA oligonucleotides used for sgRNA synthesis were designed using the CRISPOR algorithm (http://crispor.tefor.net), and sgRNAs were synthesized using the GeneArt Precision gRNA Synthesis Kit (Invitrogen, A29377). Nucleofection was carried out at 1230 V or 1300 V (four pulses of 10 ms) for U2OS or 143B cells, respectively. Following nucleofection, cells were seeded into six-well plates and allowed to recover for 48 h. For single-cell clone isolation, cells were sorted using a spectral cell sorter (Cytek Aurora CS). Genomic DNA was extracted, and a pair of primers (primer 1A, GAGTGTGCATCCGACATCCTC; primer 2A, CTCATCACCATTCTTTTGGCTG) flanking SDHA-targeted regions was used to perform PCR screening. Clones were considered positive for gene editing when the amplified product was 551 bp, indicating deletion of the whole DNA fragment between targeted regions. The absence of SDHA protein was confirmed by western blot against SDHA (Santa Cruz, sc-166909), performed following separation of total cell protein extracts on 10% SDS–PAGE gels. Positive clones were further validated by Sanger sequencing of the SDHA gene.
CRISPR–CAS9-mediated generation of DHODHKO cell line
A clonal 143B-DHODHKO cell line was generated by following a previously published protocol59, with slight modifications. Each pair of gene-specific sgRNAs (pair 1, CCGCGGAGGACTACGCAGAA; pair 2, ATAGAAACGCTCATCTCCCG) targeting different regions of the gene was cloned into a TCLV2 plasmid (Addgene, 87360)60. Lentiviral particles were made in the Viral Vectors Unit at CNIC. All 143B cells were seeded on six-well plates, and at 80% confluency, they were transduced with lentiviral particles in DMEM supplemented with only 2% FBS, which was changed the following day for fresh complete medium. After 48 h, infected cells were selected by treatment with 1 µg ml−1 puromycin in complete DMEM. After 48 h, surviving cells were treated with 2 µg ml−1 doxycycline to induce Cas9-2A-eGFP expression and were then sorted into a 96-well plate at one cell per well using a FACSAria cell sorter. Final clones were selected by observing cell death in medium lacking uridine and confirmed by running total cell protein extracts on a 12.5% SDS–PAGE gel and performing a western blot against DHODH (1:1,000) (Santa Cruz, sc166348).
Re-expression of DHODH and ScURA in DHODHKO cell line
The 143B-DHODHKO cells were used to re-express a WT version of hDHODH, a version of the protein lacking the mitochondria peptide signal and the transmembrane alpha helix (hDHODHΔMTS) and codon-optimized ScURA protein fused to a c-Myc tag (ScURA–myc). The vectors used were constructed and packaged by VectorBuilder, and the lentiviral particles were generated by the Viral Vectors Unit at CNIC. 143B-DHODHKO cells were seeded on six-well plates, and at 80% confluency, they were transduced with lentiviral particles in DMEM supplemented with 2% FBS, which was changed the following day for fresh complete medium containing uridine. Infected cells were selected by treatment with 400 µg ml−1 hygromycin B for at least 4 days. Re-expression of the proteins was confirmed by running total cell protein extracts on a 12.5% SDS–PAGE gel and performing a western blot against DHODH (1:1,000) (Santa Cruz, sc166348) and c-myc (1:5,000).
Metabolite extraction from cultured cells
For each condition, cells were seeded in six-well plates (with one plate kept aside for cell counting) in DMEM + 10% dialysed FBS, no uridine. The day after seeding, media containing inhibitors (1 μM each antimycin and myxothiazol, 2 μM brequinar) and radio-labelled tracers (2 mM [α-15N]glutamine or [U-13C]glutamine; Cambridge Isotope Laboratories) were added. After 24 h of incubation, metabolites were extracted in metabolite extraction buffer (50% methanol, 30% acetonitrile, 20% water (all liquid chromatography–mass spectrometry grade), 5 µM valine-d8, CK isotopes and DLM-488). Cells were washed with PBS, and 500 μl of cold metabolite extraction buffer per million cells was added to the wells; the plate was then immediately placed at −80 °C for 20 min. The cell lysate–extraction buffer solution mixture was shaken at maximum speed for 15 min at 4 °C, centrifuged at 13,000g for 30 min at 4 °C and the supernatant was collected in a HPLC vial. A pooled sample was prepared by taking 15 μl from each individual sample.
Liquid chromatography–mass spectrometry analysis
Metabolites were separated on a Millipore SeQuant ZIC-pHILIC analytical column (5 µm, 2.1 × 150 mm) coupled to a guard column (2.1 × 20 mm, 5 µm). The mobile phase consisted of solvent A (20 mM ammonium carbonate with 2.5 µM medronic acid, adjusted to pH 9.7 with ammonium hydroxide) and solvent B (acetonitrile in water, 95:5, v/v). The column oven was maintained at 40 °C, and the autosampler was kept at 4 °C. Chromatography was performed at a flow rate of 0.20 ml min−1 with the following gradient programme: 0–2 min, 85% B; 2–14 min, linear decrease to 30% B; 14–15 min, isocratic at 30% B; 15–17.1 min, re-equilibration to 85% B; and 17.1–23 min, hold at 85% B.
Metabolite quantification was performed on a Vanquish Horizon UHPLC system coupled to an Orbitrap Exploris 120/240 mass spectrometer (Thermo Fisher Scientific) equipped with a heated electrospray ionization source. Ionization parameters were set to +3.5 kV for positive mode and −2.8 kV for negative mode, with an RF lens setting of 70, a heated capillary temperature of 320 °C and an auxiliary gas heater temperature of 280 °C. Sheath gas flow was set to 40, auxiliary gas to 15 and sweep gas was disabled. For MS1 acquisition, data were collected in full scan mode over a mass range of m/z = 70–900, using a standard automatic gain control target with an automatically determined maximum injection time. Data were acquired with polarity switching at an Orbitrap resolution of 120,000. Untargeted metabolite profiling was performed using the AcquireX Deep Scan workflow with iterative data-dependent acquisition on pooled samples. Full scans were acquired at a resolution of 60,000, with tandem mass spectrometry fragmentation at 30,000 resolution and a minimum intensity threshold of 5.0 × 103. Dynamic exclusion was triggered after a single event (10 s, ±5 ppm), and precursors were isolated with a 1.2 m/z window. Stepped higher-energy collisional dissociation energies of 30, 50 and 150 were applied, with mild trapping enabled to improve signal quality.
Metabolite identification was performed using Compound Discoverer (v.3.2; Thermo Fisher Scientific). Compounds were assigned based on precursor m/z values within 5 ppm of the theoretical mass, fragment ion matches within 5 ppm to an internal spectral library of authentic standards analysed under identical data-dependent tandem mass spectrometry conditions (minimum match score of ≥70), and retention times within 5% of those of purified standards under the same chromatographic conditions. Peak area integration and chromatogram evaluation were carried out in TraceFinder (v.5.1; Thermo Fisher Scientific), and isotopologue distributions were corrected for natural isotope abundance using the AccuCor package61.
Semi-targeted metabolomics data analysis
Features were filtered using the 80% modified rule62, followed by exclusion of metabolites with a coefficient of variation of >30% in pooled quality control samples63. These filtering steps retain stable metabolites consistently detected in the samples, thereby preserving only robust and reproducible features. Intensities were normalized to the valine-d8 internal standard to correct for sample-to-sample technical variations, followed by LOESS signal correction to account for a systematic run order-dependent drift. Missing values were imputed with the group mean, while features entirely absent in a group were substituted with half of the minimum detected intensity in the dataset. Subsequently, data were log2-transformed to stabilize variance and improve normality before statistical analysis. Principal component analysis was performed using the prcomp function from the R stats package64, and Hotelling’s T2 test was applied to identify sample outliers using the qcc package65. Sample group differential abundance was assessed using two-sided Student’s t-tests, and P values were adjusted for multiple testing using the Benjamini–Hochberg method from the R base stats package. Finally, readxl66 and tidyverse were used for general scripting67.
Transcriptomic analysis
RNA sequencing was performed by the CNIC Genomics Unit team. RNA libraries were produced using the Illumina TruSeq RNASeq kit and sequenced in an Illumina HiSeq 2500 Sequencer. Downstream analysis was performed using the nf-core/rnaseq analysis pipeline68 using Nextflow (v.24.10.5). In brief, following quality control with FastQC, adaptor sequences were removed using Trim Galore! The cleaned reads were then aligned to the Homo sapiens reference genome (GRCh38, Ensembl release 104) using the STAR aligner. Transcript-level abundances were quantified with Salmon, and gene-level read counts were generated using featureCounts. Raw counts were normalized using the variance stabilizing transformation function, and differential gene expression analysis was performed within the DESeq2 package69. P values were adjusted for multiple testing using the Benjamini–Hochberg procedure. Pathway enrichment analysis was performed using clusterProfiler70 with statistics obtained from DESeq2. Transcription factor activity was inferred with decoupleR23 (univariate linear model), with DoRothEA regulons71. We filtered for regulons with a high level of confidence (‘A’). Generation of heatmaps and figures was done in R using the pheatmap package.
Statistics and reproducibility
Data are presented as mean ± s.d. Statistical analyses were performed using GraphPad Prism 10. The statistical tests used, sample size and post hoc corrections are indicated in the figure legends. All statistical tests were two-tailed and had an alpha of 0.05 as the significance threshold. *P < 0.05. No data were excluded.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
First Appeared on
Source link

