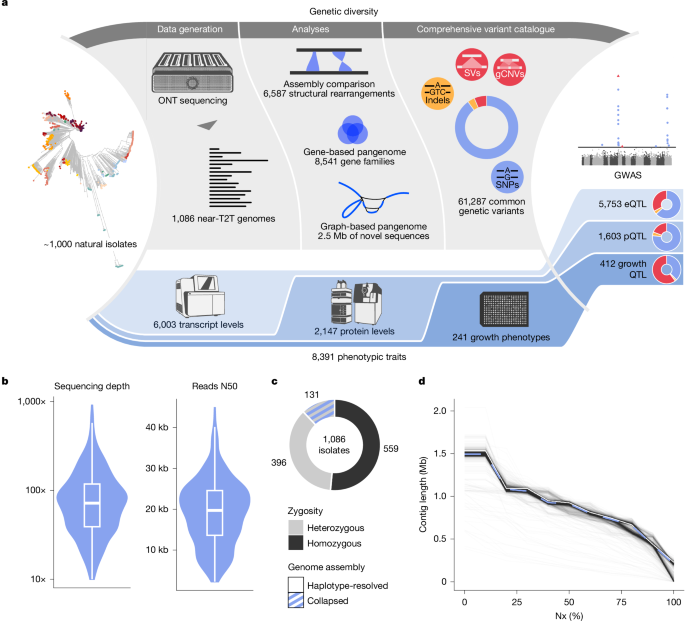
From genotype to phenotype with 1,086 near telomere-to-telomere yeast genomes
Strain culture and DNA extraction
We used a collection of S. cerevisiae isolates that were previously sequenced using short-read sequencing19. For each isolate, we obtained single colonies from frozen stock on solid YPD (1% yeast extract, 2% peptone, and 2% glucose) and cultured one colony per strain in 25 ml of liquid YPD at 30 °C under shaking (120 rpm). After the culture reached saturation (approximately 1.5 days), the nuclear DNA was extracted from the cells using either a previously described protocol69 or a Monarch HMW DNA Extraction Kit (New England Biolabs). The cells from the saturated culture were treated for 2 h with zymolyase (1,000 U ml−1) in 1 M sorbitol to produce spheroplasts. The spheroplasts were then processed with the Monarch HMW DNA Extraction Kit. Samples with a DNA concentration higher than 30 ng µl−1 were retained for DNA sequencing.
Sequencing data
Long reads sequencing data were obtained using Oxford Nanopore sequencing technology. The library was prepared according to the following protocol, using the Oxford Nanopore SQK-LSK109 and SQK-LSK114 kits. Genomic DNA fragments were repaired and 3′-adenylated with the NEBNext FFPE DNA Repair Mix and the NEBNext Ultra II End Repair/dA-Tailing Module (New England Biolabs). Sequencing adapters provided by Oxford Nanopore Technologies (Oxford Nanopore Technologies) were then ligated using the NEBNext Quick Ligation Module (NEB). After purification with AMPure XP beads (Beckmann Coulter), the library was mixed with the sequencing buffer (ONT) and the loading bead (ONT) and loaded on PromethION R9.4.1 and R10.4.1 flowcells. Basecalling was performed with guppy 5.0.16 (https://nanoporetech.com). To confirm the correspondence of novel long-read sequences with previously generated short-read sequences, we compared SNPs inferred from both types of data. Long and short reads were mapped independently on the reference genome using minimap2 v.2.24 (ref. 70) and bwa-mem2 v.2.2.1 (ref. 71), respectively, and SNPs were inferred with longshot v.0.4.5 (ref. 72) and gatk v.4.5.0.0 (ref. 73). The reference genome version R64-3-1 was downloaded as a fasta file from the Saccharomyces genome database74 website (https://www.yeastgenome.org). We computed the pairwise distance between all samples based on short reads and long reads SNPs using plink v.1.9 (ref. 75). Cases with unclear correspondence between short and long reads were discarded.
Reads phasing
Sequencing data from non-polyploid heterozygous samples with coverage higher than 20x were phased to obtain one read set for each haplotype. Long reads were mapped on the reference genome using minimap2 v.2.24 (ref. 70) with the option -ax map-ont. SNPs were called with longshot v.0.4.5 (ref. 72) –no_haps –min_cov 7 –min_alt_count 7 –min_alt_frac 0.2. Regions of loss of heterozygosity, defined as 50-kb windows containing fewer than 10 SNPs, were detected and removed from the phasing process. SNPs were phased using whatshap phase v.1.4 (ref. 76), and each sequencing read was tagged HP1, HP2 or unassigned. Unassigned reads were downsampled at 50% coverage with filtlong v.0.2.1 (https://github.com/rrwick/Filtlong) –min_length 1000 –length_weight 10 –keep_percent 50 –min_mean_q 9 to maintain similar coverage to the phased reads. Read set for each haplotype was finally obtained by combining phased reads and unassigned reads.
Genome assembly
The genome assembly pipeline (Supplementary Fig. 2) was run with raw sequencing data of 1,027 samples with more than 10x sequencing coverage, in addition to phased sequencing data for 433 non-polyploid heterozygous samples. Sequencing data were systematically downsampled to 30x using filtlong –min_length 1000 –length_weight 10 –target_bases 360000000 –min_mean_q 9 and additionally to 40x when raw coverage was higher than 40x with –target_bases 480000000. Raw and downsampled sequencing reads were assembled with 3 genome assemblers: (1) Necat v.0.0.1_update20200803 (ref. 77); (2) Flye v.2.9 (ref. 78); and (3) SMARTdenovo -c 1 (ref. 79) using reads cleaned with Necat. Redundancy within each genome assembly was removed by discarding contigs covered on more than 95% by other contigs of the draft assembly. Nuclear contigs were then selected by sequence similarity with a database of S. cerevisiae nuclear chromosomes built from 142 genome assemblies7, discarding chromosomes containing mitochondrial insertions. For each sample and each phased haplotype, the best genome assembly was selected with seven criteria chosen to favour completeness and contiguity: (1) each genome assembly must cover the reference genome over 95% of its length; (2) cover at least 80% of each reference chromosome (except for chromosome 1 for which the threshold was lowered to 75% because of a more variable size); (3) does not cover more than 50% of the mitochondrial genome; (4) does not contains fused chromosomes, identified as contigs containing multiple centromeres; (5) favours the lowest number of contigs required to cover 95% of the reference genome; (6) favours the lowest number of contigs; and finally (7) favours the largest total length. The second-best genome assembly, obtained with a different assembler, was also kept for further utilization in the SV detection pipeline. Genome assemblies were then polished with both long reads using medaka consensus -m r941_prom_sup_g507 1.8.0 (https://github.com/nanoporetech/medaka) and Illumina short reads using HapoG 1.3.3 (ref. 80). For phased haplotypes, contigs of each haplotype were concatenated to perform the short reads polishing. Finally, scaffolding against the reference genome was performed using ragout –solid-scaffolds 2.3.1 (ref. 81). For cases for which the scaffolding generated fused chromosomes, the non-scaffolded genome assembly was retained. Assembly contigs were named and ordered according to their sequence similarity to reference chromosomes. For strain XTRA_FHL, whose sequencing data was contaminated by a Kluveromyces marxianus isolate, K. marxianus contigs were manually removed.
Quality assessment and genome annotation
Correctness of genome assemblies was evaluated with Merqury82, and completeness was assessed with miniBusco37. Gene prediction and detection of TEs, centromeres and subtelomeric elements were performed through the LRSDAY pipeline v.1.7.0 (ref. 83). Telomeric sequences were identified across all assemblies using Telofinder7.
SV detection
SVs were detected by individually comparing the generated assemblies with the reference genome (SGD R64 genome assembly of strain S288c, GenBank ID: GCA_000146045.2) using MUM&Co v.3.8 (ref. 84) with the -g 12000000 option. SV calling was run on 1,482 genome assemblies from 1,086 isolates (including 396 isolates with phased genome assemblies). The pipeline uses whole-genome alignments obtained via the MUMmer4 software85 to detect insertions, deletions, duplications, contractions, inversions, and reciprocal translocations exhibiting a size larger than 50 bp. To be validated, SVs had to be detected in at least two independent assemblies. To avoid removing singleton SVs—that is, those present in a single haplotype—we considered an additional set of 1,329 ‘second-best assemblies’ for 959 isolates of our collection, obtained from an alternative assembler and that met the completeness quality threshold defined. A total of 2,811 single sample VCF files were obtained and merged into a single multisample VCF file. First, insertions, deletions, duplications, contractions, and inversions were merged using Jasmine v.1.1.5 (ref. 86), which is based on an SV proximity graph that consider SV breakpoint position and length. Given that Jasmine’s algorithm does not consider two breakpoints for a single SV, a custom merging strategy was developed for translocations, as these involve two distinct breakpoints in the genome. This strategy is based on the construction of a translocation graph, linking pairs of translocations with both breakpoints within a 10 kb region. Each connected component of this graph was treated as a single SV and appended to the Jasmine’s output.
To minimize false positives, we retained only SVs detected in at least two genome assemblies, coming from different isolates, haplotypes, or genome assemblers. We then discarded the second-best assemblies from the VCF file. Finally, phased haplotypes from the same isolate were merged into phased heterozygous genotypes, resulting in the final SV VCF file containing 1,086 samples. For further analyses, we classified insertions and deletions as presence–absence variants (PAVs), and duplications and contractions as CNVs, to avoid reference-biased terminology. Therefore, PAVs and indels are only distinguished by their sizes—higher or lower than 50 bp, respectively.
Detection of Ty-related SVs
The sequence of PAVs, CNVs and inversions were aligned to a Ty retrotransposon database using blast v.2.12.0 (ref. 87) with the -dust no -perc_identity 95 options. The database was constructed from the sequences of the 48 Ty elements present in the reference genome in addition to the 4 solo LTRs sequences. SVs with more than 50% of their length covered were defined as Ty-related SVs.
Structural diversity
To quantify structural diversity, we adapted the classical formula for pairwise nucleotide diversity88 to account for SVs. We defined the structural diversity \({\pi }_{{\rm{SV}}}\) as the average number of structural differences per site between two sequences within a population:
$${\pi }_{{\rm{SV}}}=\frac{n}{n\,+\,1}\sum _{ij}{x}_{i}{x}_{j}{\pi }_{ij}=\frac{n}{n\,+\,1}\mathop{\sum }\limits_{i=2}^{n}\mathop{\sum }\limits_{j=1}^{i-1}2{x}_{i}{x}_{j}{\pi }_{ij}$$
where \({x}_{i}\) and \({x}_{j}\) are the frequencies of haplotypes \(i\) and \(j\), \({\pi }_{{ij}}\) is the number of SV differences between the haplotypes i and j and \(n\) is the total number of haplotypes. Each SV, including PAVs, CNVs, inversions and translocations, was treated as a discrete event, regardless of its size. An SV was considered present in a window if it overlapped the window by at least 1 bp for PAVs, CNVs, and inversions, or if a translocation breakpoint fell within the window.
We computed \({\pi }_{{\rm{SV}}}\) for each type of SV individually on 10-kb sliding windows (1 kb step). Outlier regions were defined as regions with \({\pi }_{{\rm{SV}}}\) greater than the third quartile plus 5 times the IQR for PAVs and translocations, plus 10 times the IQR for inversions, and plus 20 times the IQR for CNVs, to account for baseline variability. To detect regions associated with specific clades, we tested for the over-representation of SVs located in the region of interest in each clade using a two-sided fisher’s exact test with FDR correction.
SNP and indels detection
SNPs and indels were detected for the 1,086 isolates based on the alignment of paired-end Illumina reads7,19,36 to the reference genome. The reads were mapped to the reference genome using bwa-mem2 mem v.2.2.1 (ref. 71) with default parameters and samtools sort v.1.15.1 (ref. 89). The HaplotypeCaller command from gatk v.4.2.3.0 (ref. 73) was used with option –emit-ref-confidence GVCF to generate single sample GVCF files. These files were then gathered into a single multisample vcf file using commands GenomicsDBImport and GenotypeGVCFs –include-non-variant-sites, following gatk’s germline short variant discovery workflow (https://gatk.broadinstitute.org/hc/en-us/articles/360035535932-Germline-short-variant-discovery-SNPs-Indels). Low-quality genotypes (DP < 10 and GQ < 20) were set to missing using bcftools v.1.18.1 (ref. 89) with the +set-gt command. Sites with fewer than 99% informed genotypes and sites exhibiting excess of heterozygosity (ExcHet > 0.99) were removed. Finally, SNPs and indels were separated into two vcf files using bcftools, and complex loci spanning both SNPs and indels were discarded.
Neighbour-joining trees
Neighbour-joining trees were constructed independently from SNPs and SV matrices (1,474,884 and 6,587 markers, respectively, for 1,086 isolates) using the R packages ape90 and SNPRelate91.
Site frequency spectrum
Annotations of SNPs and indels were obtained using SnpEff v.5.1 (ref. 92) with the -no-downstream -no-upstream options.
Comparison of the number of SVs and SNPs per isolate
We used a simple linear regression to model the relationship between the number of SNPs and SVs in each isolate. We further used the R package chisq.posthoc.test93 to test for clades and super clades that deviated from the linear relationship between the number of SNPs and SVs. The chisq.posthoc.test function was used with a matrix containing the mean number of SNPs and SVs for each clade, with the method = ‘bonferroni’ option.
Gene-based pangenome
The gene-based pangenome was built on the de novo annotated coding sequence (CDS) of genomes with a Merqury quality value (QV) superior to 40, as a lower QV is associated with an increased number of singleton gene families (present in a single isolate), potentially being false positive CDS (Supplementary Fig. 11a). After the Merqury QV filtering, we considered 762 genomes corresponding to 651 isolates (Supplementary Fig. 12) for the construction of the gene-based pangenome (Supplementary Fig. 13).
First, we transferred the annotation of the reference CDS on the CDS identified de novo in the assemblies with a nucleotide sequence similarity search, using blastn v.2.12.0 (ref. 87) with options -dust no -prec_identity 95 -strand plus. The reference annotations version R64-4-1 were downloaded from the Saccharomyces genome database74 website (https://www.yeastgenome.org) in gff3 format. For each pair of de novo and reference CDS, the annotation was transferred when one of these cases was true: (1) the de novo CDS is covered by the reference CDS on more than 90% of its length; (2) the reference CDS is covered by the de novo one on more than 50% of its length; (3) the reference CDS is covered by the de novo one on more than 30% of its length and both start and end of the reference CDS are covered (that is, the alignment spans the first and last 10 bp of the CDS). De novo CDS covering less than 80% of the reference sequence have been annotated as truncated. Additionally, de novo CDS were annotated as Truncated when their reference homologue was covered on less than 80% of their length and alignment did not cover the start and end of the CDS. However, this additional information is purely informative and was not considered for the pangenome construction. We then filtered out all the genes with a length inferior to 100 bp.
Second, we identified gene families with a graph-based strategy. We ran a nucleotide sequences similarity search on the 6,673 reference CDS (larger than 100 bp) and 77,322 de novo CDS for which no annotation was transferred in the previous step, using blastn with the same options mentioned before. A graph was then built using CDS as nodes and sequence homology as edges, using the python package NetworkX94. Homology was considered when an alignment between two CDS covered both at 50% of their length or either at 90%, or when a reference annotation was transferred. Connected components with a density lower than 0.4 were further split into Louvain’s communities, and each component or community was then considered as a gene family. For each gene family, a representative sequence was chosen as the reference CDS with the highest degree in the family, or the de novo CDS with the highest degree when no reference CDS was present. Finally, CDS with less than 100 bp of unique sequence in the pangenome (that is, fragments of sequences strictly identical between multiple CDS) were iteratively removed. Identical matches within the pangenome were identified using blastn with options -dust no -strand plus -wordsize 100 -penalty -10000 -ungapped.
Third, to estimate gene presence/absence within the 1,086 isolates, we used a sequencing depth-based approach. Illumina reads were mapped to the representative CDS of each gene family using bwa-mem2 mem v.2.2.1 (ref. 71) with options -U 0 -L 0,0 -O 4,4 -T 20, which remove penalty for unpaired reads, reduce penalty for reads clipping and gap opening and lower the minimum score required. These relaxed parameters were chosen to prevent mapping issues due to diversity within gene families. Read depth over each CDS was calculated using samtools depth v.1.16.1 (ref. 89) with option -aa. For each CDS in each isolate, a normalized depth was computed as the ratio of the median depth of the CDS (discarding non-unique fragment identified in the previous step) over the median depth of all sequences. Because a given normalized depth could have different meaning according to the ploidy of the isolate, we adjusted the normalized depth with the ploidy and a correcting factor x:
$${\rm{Gene}}\;{\rm{presence}}\propto {{\rm{Normalized\; depth}}\times {\rm{p}}{\rm{l}}{\rm{o}}{\rm{i}}{\rm{d}}{\rm{y}}}^{x}$$
To identify the optimal value of x, we built a gold standard gene presence/absence matrix considering 553 isolates and 5,792 genes for which both de novo annotated genome assemblies and transcriptomic data20 were available. A gene was considered present in an isolate when it was: (1) annotated in the genome assembly; and (2) expressed at ≥2 transcripts per million (TPM) in the corresponding RNA-seq data. A gene was considered absent when it was: (1) not annotated; and (2) had expression <2 TPM. All other cases were excluded to avoid ambiguity. This gold standard was solely used to evaluate the accuracy of gene presence/absence calls from sequencing depth, by generating a precision-recall curve for various values of x (Supplementary Fig. 11b). The optimal performance (highest area under the precision-recall curve; Supplementary Fig. 11c) was achieved with x = 0.15. We further computed the precision and recall values using different thresholds (Supplementary Fig. 11d) and chose a threshold of 0.3 as the recall declined sharply beyond this point. Using this threshold, we estimated the presence of all the gene families of the pangenome across the 1,086 isolates.
Importantly, expression data was used solely to calibrate the sequencing depth-based model, and was not used to call gene presence or absence in any genome. Final gene presence/absence calls were made entirely from DNA sequence data using mapped short-read depth.
Pangenome annotation
We sought to annotate the origin and function of novel genes with protein sequences similarity search against a curated database. We built a blast database by coupling the RefSeq protein database with a custom database containing Fungi protein sequences from Shen et al.95 and S. paradoxus protein sequences from Yue et al.96. The sequence similarity search was run using blastp87 with default parameters, and the obtained results were further filtered with a minimum protein identity of 30% and a minimum query coverage of 50%. We categorized the origin of each novel gene as: (1) fast-evolving gene when the best hit was a S. cerevisiae protein; (2) introgression when the best hit was a Saccharomyces protein other than S. cerevisiae; (3) HGT when the protein came out of the Saccharomyces genus; and (4) unknown when no sequence similarity was found. We also transferred the gene ontology (GO) terms associated with the best protein hit in RefSeq to each novel gene (using the same identity and coverage filters as above) and inferred the GO terms of the whole pangenome based on sequence using InterProScan v.4.65-97.0 (ref. 97).
Transcriptomics
Reads were mapped on the CDS of the pangenome using STAR v.2.7.9.a98 with default parameters. Number of reads mapped to each gene family was retrieved using samtools idxstats v.1.16.1 (ref. 89) and TPM were computed with a custom python script.
Rarefaction curves
For both SVs and gene families, rarefactions curves were obtained using the R package iNEXT v.3.0.0 (ref. 99). The iNEXT function was used with a presence–absence matrix of SVs or genes and the options datatype = ‘incidence_raw’ and k = 400. We interpreted the species richness as the total number of SV or gene families in the species and the sample coverage estimate as the species coverage. For the core genome, we first used a matrix of missing genes (filled with 1 when the gene was absent and 0 when the gene was present) as input of the iNEXT function, with the same parameters as before. We then subtracted the rarefaction obtained from the missing genes to the species estimate of the pangenome (8,583 genes), to obtain the rarefaction of the core genome. Finally, we obtained the rarefaction of the accessory genome by subtracting the core genome rarefaction to the pangenome rarefaction.
Clade-specific variants
Clade-specific SVs and genes were obtained using a simple-over-representation analysis based on hypergeometric tests. We used the fora function from the fgsea R package v.1.27.0 (ref. 100) using the list of clades and super clades as ‘pathways’, the isolates having the SV or gene as ‘gene’ and the total list of isolates as ‘universe’. The function was run for each SV or gene present in at least two isolates, except SV or genes present in all isolates. Results were further concatenated, and P value were adjusted using the FDR method.
GO analysis
We used the GO annotations of the reference genes available from SGD (https://current.geneontology.org/annotations/sgd.gaf.gz), in addition to the transferred GO terms for the novel genes (see ‘Pangenome annotation’) and the GO terms inferred with InterProScan97 for the entire pangenome. We discarded terms with a size larger than 500, as well as terms with a size smaller than 2 (except for terms with a reliable evidence code—that is, inferred from mutant phenotype (IMP), inferred from direct assay (IDA) and inferred from genetic interaction (IGI)). For some analyses, we performed a GO term semantic similarity reduction using the calculateSimMatrix and reduceSimMatrix(threshold = 0.7) functions of the rrvgo R package v.1.10.0 (ref. 101). GO term enrichments were performed using the fora function from the fgsea v.1.27.0 R package100. Clade-specific GO terms were detected similarly to clade-specific genes.
Construction of an exhaustive genotype matrix
To build the most comprehensive genotype matrix possible, we combined SNPs and indels with SVs called from both genome assembly comparisons and gene-based pangenome construction. We transformed the normalized depth computed for each CDS–isolate pair (see ‘Gene-based pangenome’) into biallelic variants by setting multiple depth thresholds (starting from 0.25 and increasing by steps of 0.5, as we would expect for a diploid isolate). We discriminated isolates having a normalized depth below or above each threshold for each CDS. In that way, we capture both the presence–absence of each CDS in the population, in addition to the variation in copy number. The complete loss of a gene was considered as deletion.
Although combining multiple strategies for SV calling ensures the comprehensiveness of our variant catalogue, it results in redundancy from the segmental SV detections (that is, assembly comparisons) and the gene-based CNV and deletion detection. For example, a deletion spanning three consecutive genes would result in one segmental deletion and the deletion of each individual gene. The four SV records would exhibit high linkage disequilibrium as they all correspond to a single event. Additionally, aneuploidies are captured by CNVs of all genes present on the aneuploid chromosome. Although it corresponds to a single event, it results in many gene–CNV records, that exhibit high linkage disequilibrium with each other. This redundancy can be easily removed with linkage pruning to prevent duplicated genetic associations in further analyses.
Heritability estimates
Heritability estimates were computed for complex traits, defined as traits having no association with a P value lower than 1 × 10−20 by GWAS, as high effect predictors may bias the estimations. LDAK v.4.2 (ref. 102) was used for the computation. Phenotypes were normalized using a rank-based inverse normal transformation. Plink matrices of SNPs, indels and SVs were first used to generate independent kinship matrices using the LDAK thin model. Weights for each variant were first computed using LDAK with arguments –thin –window-prune .98 –window-kb 20, and kinship were generated using arguments –calc-kins-direct –weights –power -.25. Trait heritability was then estimated using all three kinship matrices together (–mgrm option), using ploidy as covariate and the option –constrain YES to ensure positive values of heritability.
GWAS
We ran GWAS using a linear mixed model implemented in FaST-LMM v.0.4.6 (ref. 57). Phenotypes were normalized in the same way as for heritability estimates. SNPs, indels and SVs were filtered for MAF at 5%. This MAF filtering retained 89,906 SNPs (6.4% of all SNPs), 2,415 indels (4.3%) and 7,708 SVs (10.7%). These allele frequency differences were not accounted for. Genotypes were pruned for linkage disequilibrium using plink v.1.9 (ref. 75) with option –indep-pairwise 50 kb 1 0.8, yielding 54,544 SNPs, 2,203 indels and 4,540 SVs. Variants were further combined in a single plink matrix, which was used as both kinship and test set for GWAS. To preclude the effect of ploidy and aneuploidies on the genetic associations, both were added as covariates. Ploidy was encoded as a numerical covariate and aneuploidies were encoded for each chromosome as −1, 0 or 1, representing loss, expected copy number or gain, respectively. To correct for the large number of variants tested, a trait-specific P value threshold was defined using a permutation test with 100 permutations and alpha = 0.05. In brief, for each trait, associations were run 100 times on permutated phenotypes retaining the lowest P value for each run. The P value threshold corresponds to the 5% quantile (that is, the fifth-lowest P value across permutations), corresponding to an FDR correction of 5%.
Local variants were defined as located in a 25-kb region around the gene of interest or linked to a pruned variant located in this region. For translocations, both breakpoints were considered for the definition of local variants. To further account for linkage disequilibrium between associated variants, groups of linkage were identified, defined as connected components of variants associated with a same trait and in linkage disequilibrium (based on a 0.5 r2 threshold and a maximal physical distance of 50 kb). For each group, only the variant with the lowest P value was retained, leading to the final number of 4,564 QTL.
Graph construction and novel sequence detection
We build a graph pangenome using the Minigraph-Cactus pipeline v.2.6.4 (ref. 59) with 500 haplotypes, including the reference genome and 499 genomes selected to represent a maximum number of SVs. We used the first graph generated by Minigraph58, that uniquely contains SVs, in order to identify repetitive reference segments in the graph and novel sequences. Only segments larger than 100 bp were considered for these analyses. The segments were mapped to the reference genome using minimap2 -ax asm5 v.2.24 (ref. 70) and the coverage depth along the genome was retrieved using samtools depth v.1.16.1 (ref. 89). To determine the fraction of the graph corresponding to TEs, the segments were aligned on a Ty sequence database constructed from the previous analyses of 1,011 genomes103, using blastn v.2.12.0 (ref. 87), with option -perc_identity 70. The same alignment was performed on each of the genome assemblies used for the graph pangenome construction. Additionally, we sought for sequence redundancy in the graph using blastn -perc_identity 95 and applied a 50% coverage threshold. We built a sequence similarity graph and selected a single representative segment for each connected component. Components containing no reference segments were considered as novel sequences. Novel segments were considered as introgression when they show sequence similarity higher than 95% on a database composed of Saccharomyces genomes (GCF_000292725.1, GCF_001298625.1, GCF_002079055.1, GCF_947241705.1, GCF_947243775.1, GCA_002079085.1, GCA_002079115.1, GCA_002079145.1, GCA_002079175.1) using blastn -perc_identity 95.
Variant calling using the graph
The pangenome graph (gfa format) was converted to gbz using the vg toolkit v.1.54.0 (ref. 104) with the vg autoindex command, and snarls were detected using vg snarls. Illumina reads of 3,039 isolates with publicly available sequencing data were mapped on the graph using vg giraffe105 with options –fragment-mean 350 –fragment-stdev 100 -b fast. Gam files were converted using vg pack with option -Q 5 to remove reads with low mapping quality. We performed variant calling using vg call106 with options –genotype-snarls –all-snarls –snarls –ref-sample S288c to obtain single sample vcf files. Calling from the graph pangenome worked for 2,874 out of 3,039 isolates, the remaining ones being discarded because of an aberrantly long computing time. Single sample vcf files were merged into a single multisample vcf using bcftools merge v.1.16.1 (ref. 89). Variants supported by less than two reads were set to missing using bcftools +setGT — -t q -n. -e ‘FMT/DP > = 2’. The resulting vcf was further trimmed for non-present alternate alleles with bcftools view –trim-alt-alleles and variants were atomized into multiple ones with bcftools norm –atomize –atom-overlap “.” –multiallelics +any. The difference of length between alternative and reference alleles were used to classify alleles as SNPs, indels of SVs. SNPs show no length difference, indels have length differences inferior to 50 bp and SVs show length differences larger or equal to 50 bp.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
First Appeared on
Source link

