Mapping Plasmodium transitions and interactions in the Anopheles female
Rearing of Anopheles mosquitoes
A. gambiae (G3 strain), A. stephensi (SDA-500), and recently colonized A. coluzzi41 mosquitoes were reared in an insectary maintained at 27 °C, 70–80% relative humidity, on a 12 h:12 h light:dark cycle. Larvae were fed on fish food and adults were provided with 10% w/v glucose solution ad libitum. Adult mosquitoes were fed with purchased human blood (Research Blood Components) to induce egg laying.
dsRNA production and microinjection
PCR fragments of eGFP control (495 bp) and EcR (AGAP029539; 435 bp) were amplified from the plasmids pCR2.1-eGFP and pCR2.1-EcR as previously described51. PCR product was used to transcribe dsRNA using the Megascript T7 transcription kit (Thermo Fisher Scientific), per the manufacturer’s protocol. The resulting dsRNA was purified via phenol-chloroform and diluted to a concentration of 10 µg µl−1. Microinjections were performed as detailed previously24, with adult mosquitoes, randomly assigned to each group, being injected one day post-eclosion and given an infectious blood meal 3 days post-injection.
EcR knockdown confirmation
Resulting knockdown confirmation post-injection was performed as previously described24. Pools of 6–8 infected female mosquitoes were aspirated at 24 hpi and transferred to TRI reagent (Thermo Fisher Scientific). RNA was extracted, DNase treated, quantified and reverse transcribed. cDNA from four biological replicates was diluted tenfold and run in duplicate for quantitative PCR with reverse transcription. Gene-specific primers were used, and relative quantification was performed with the ribosomal gene Rpl19 as the reference52 (Supplementary Table 8). RNA extraction from one replicate of the single-cell experiments failed and were thus repeated with samples collected at 2 dpi. For phenotypic EcR knockdown confirmation, individual mosquito ovaries were also collected 7 dpi in 80% ethanol for later manual egg counts.
Culturing of P. falciparum parasites
P. falciparum strains used in this study include: (1) NF54 originally provided by C. Barillas-Mury from BEI Resources (MRA-1000); (2) NF54_H, highly infectious to mosquitoes, from the laboratory of M. Delves; (3) an artemisinin-resistant, Cambodian field isolate ART29 (ref. 53); and (4) a Burkina Faso polyclonal isolate P5 (refs. 25,35). All strains were identified as P. falciparum parasites by nested PCR or whole genome sequencing, and confirmed mycoplasma free. Asexual stages of parasites were cultured at 37 °C between 0.2 and 2% parasitaemia in human erythrocytes at 5% haematocrit (Interstate Blood Bank) using RPMI medium 1640 supplemented with 25 mM HEPES, 10 mg l−1 hypoxanthine, 0.2% sodium bicarbonate, and 10% heat-inactivated human serum (Interstate Blood Bank) under a gas mixture of 5% O2, 5% CO2, balanced N2. Stage V female and male gametocytes were induced by raising parasitaemia over 4% and incubating cultures for 14–20 days with daily medium change.
P. falciparum infections of Anopheles mosquitoes
Female mosquitoes were blood-fed on a mix of P. falciparum gametocyte culture, human red blood cells and human serum (ratio 1:3:6) via heated membrane feeders 3–5 days post-eclosion, and introduced into a custom-built glove box (Inert Technology) as previously described24. Unfed mosquitoes were removed, and remaining mosquitoes were kept on 10% glucose solution ad libitum, with added ATC or compound (see below). At dissection time points, mosquitoes were aspirated into 80% ethanol, frozen for 10 min and transferred to phosphate-buffered saline (PBS) on ice.
Generation of single-cell suspension
Eighty midguts from P. falciparum-infected mosquitoes injected with dsGFP or dsEcR were dissected at 36 hpi, 2 dpi, 4 dpi and 7 dpi across 4 biological replicates in ice-cold PBS. Any remaining blood meal was removed before storing the dissected midguts in ice-cold PBS. Dissections were completed within 1 h to preserve the viability of parasite and mosquito cells. Midguts were pooled into 200 µl of PBS with 1 mg ml−1 elastase (Sigma-Aldrich E7885) and 1 mg ml−1 collagenase IV (Thermo Fisher Scientific 17104019), and incubated at 30 °C, 300 rpm for 30 min in a shaking heat block. To facilitate the isolation of single cells, samples were disrupted every 10 min by pipetting with low protein-binding tips.
Samples were assessed using trypan blue staining to quantify live, single midgut cells during protocol optimization. This included systematically testing variables such as digestion time, temperature, tissue preparation (intact or minced gut) and types of pipette tip. On average, approximately 400 live, single cells were consistently recovered per mosquito midgut, while many cells remained in clumps. Given the low number of parasites (~20–30) relative to mosquito cells (~5,000) in a midgut, rather than further optimizing for complete dissociation of the midgut, we chose to enrich the parasite-to-mosquito cell ratio by removing cell clumps as they were composed mostly of midgut cells. Therefore, the cell suspension was sequentially filtered through 70 µm, 40 µm and 20 µm cell strainers (pluriSelect), with each strainer being immediately washed with 200 µl of 1× PBS to collect any adherent cells. The 20 µm cell strainers were not used for the 7 dpi samples, as oocysts at this time point are, on average, over 25 µm in diameter24. Based on the final dataset, this approach yielded an approximate 1:16 parasite-to-mosquito cell ratio. Given an estimated 5,000 midgut cells and 30 oocysts per gut—an expected ratio of about 166:1—our protocol represents a tenfold enrichment of parasites.
We chose not to collect samples beyond 7 dpi, as oocysts larger than 30 µm may clog the 10x Chromium chip. We chose not to use fluorescence-activated or magnetic-activated cell sorting as they both lengthen the isolation protocol and cause stress, reducing cell viability. The final single-cell suspension was pelleted, resuspended in 20 µl of PBS, and cell concentration and viability were determined by haemocytometer count with trypan blue staining.
Single-cell library preparation and sequencing
Single-cell libraries were prepared following the Chromium Next GEM Single Cell 3′ Reagents Kit v.3.1 (Dual Index) User Guide (RevC). In brief, single-cell suspensions were diluted to target a recovery of 4,000 cells per sample, then mixed with reverse transcription reagents and barcoded gel beads. The mixtures were loaded into wells containing partitioning oil on a 10x Chromium chip to generate single-cell emulsions. Samples from dsGFP and dsEcR mosquitoes collected at the same time point were loaded into two separate wells on the same chip. In total, 16 chips were used across 4 time points and 4 biological replicates. Within each emulsion droplet, individual cells were lysed, mRNA molecules were captured, and cDNAs were generated with cell-specific barcodes. Barcoded cDNAs were pooled, then amplified, fragmented and purified using SPRIselect, followed by ligation with Illumina adaptors and sample-specific barcodes. Each sample was assessed using an Agilent Bioanalyzer to ensure library integrity and quantified by qPCR using i7 and i5 Illumina primers, following the protocol from Universal Kapa Library Quantification Kit (Roche). The cDNA libraries from each sample were then mixed in equal proportions, spiked with 1% phage cDNA (PhiX, Illumina). The proportion of parasite reads relative to mosquito reads was estimated using an initial Illumina iSeq 100 run, which showed that around 0.05%, 0.1%, 0.6% and 1.3% reads mapped to parasites at 36 hpi, 2 dpi, 4 dpi and 7 dpi, respectively. To obtain sufficient coverage of parasite transcripts, final sequencing was performed across all lanes of two NovaSeq S4 flow cells (Broad Institute), with an expectation of 1,974–51,316 reads per parasite ranging from 36 h to 7 dpi (see Supplementary Note for detailed calculation).
scRNA-seq data processing and analysis of P. falciparum
FASTQ files from each sample originating from different flow cells were concatenated and mapped to the genomes of P. falciparum 3D7 (PlasmoDB.org, v.58)54 using 10x Cell Ranger software v.7.0.1 (ref. 55). The resulting count matrix for each sample was processed and filtered using Scanpy (v.1.9.1) in Python (v.3.10)56. Based on the individual sample profile, low-quality cells, dead cells and empty droplets were removed according to the number of reads (unique molecular identifier (UMI) count), number of genes (gene count) and proportion of mitochondrial reads per cell (Supplementary Table 1). The count matrix from each sample was merged into a single AnnData object by Scanpy, and doublets were removed running Scrublet software in Python before conversion to Seurat using SeuratDisk’s function Convert57. After quality control, the average number of reads per cell ranged from 649 at 36 hpi to 9,043 at 7 dpi. Pseudobulk differential expression analyses were initially conducted treating the samples as independent observations for comparison. The pseudobulk count matrix was generated for each sample by summing raw gene counts across all cells in Python with Pegasus. Several comparisons were conducted—between each time point in parasites from dsGFP-injected mosquitoes as well as between parasites from dsEcR– and dsGFP-injected mosquitoes of the same time point—using a Python wrapper for the DESeq2 package in R58. Significance was set as log2 fold change greater than 1 or less than −1, and an adjusted P value below 0.05. Functional enrichment analysis of upregulated or downregulated gene lists was performed using GoProfiler59.
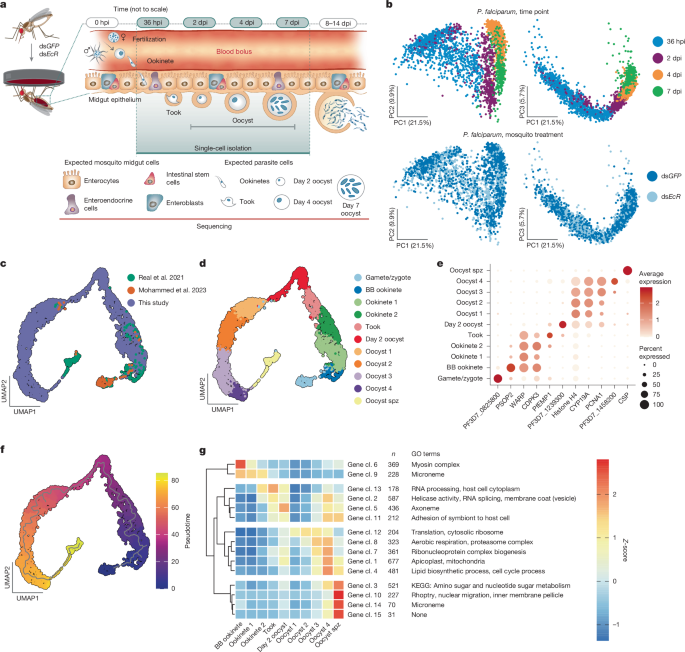
A standard single-cell analysis pipeline was then performed with Seurat and dependent packages in R 4.3.2 in RStudio 2023.9.1.494 (ref. 60). Highly variable genes were identified with the FindVariableFeatures function before data were scaled (ScaleData, default settings) and a PCA plot was generated based on these features only. Next, high-quality parasite cells were integrated with existing datasets, including blood bolus ookinetes at 24 hpi and 48 hpi, gametes, zygotes and ookinetes from 2–20 hpi, and sporozoites released from oocysts21,22 at 12 dpi. In brief, Seurat objects (v.5) were created for external datasets and reads were normalized to 10,000 transcripts, before the three Seurat objects were merged. Variable features were identified with default parameters of the FindVariableFeatures function, the data were scaled (ScaleData), and PCA was performed on the first 50 dimensions (RunPCA, default settings). Integration to correct for batch effect and differences in single-cell methodology was conducted using RPCA, which is the preferred approach when minimal overlap between datasets is expected. Subsequently, a UMAP was generated including the top 12 principal components through the RunUMAP function, with parameters set to min.dist = 0.4, and repulsion.strength = 2. Cell clusters were defined by the Louvain algorithm at a resolution of 0.5. Cluster markers were identified using the FindAllMarkers function, focusing only on positive markers with a log2 fold change above 1. Genes with FDR values below 0.01 were utilized to label each cluster.
The Seurat object was converted into a cds object in Monocle3 (ref. 61), and the trajectory analysis was performed using the learn_graph function (close_loop = F, ncenter=500). Pseudotime analysis was conducted by setting the node in the gamete–zygote cluster as the start point. Gene network analysis focused on our data and was performed using the graph_test function (neighbor_graph = “principal_graph”), to identify co-expressed genes that change as a function of pseudotime (FDR < 0.05). Co-expressed genes were further clustered by the find_gene_modules function with a resolution of 0.0023. The expression of all genes within each gene cluster was aggregated using the aggregate_gene_expression function. Enriched GO terms were determined by analysing all the genes within a gene cluster in GoProfiler59. Differential expression of protein coding genes was compared between dual-read parasites at 36 hpi and parasites that do not share a barcode with a mosquito cell, binning for parasite cluster, using logistic regression. A subsequent analysis focused on dual-read parasites only at 36 hpi, and compared those interacting with an ISC/EB to the remainder.
Mosquito scRNA-seq data processing and analysis
All FASTQ files from the different sequencing runs were concatenated and mapped to the A. gambiae PEST genome (VectorBase.org, v.58)54, using the 10x Cell Ranger software v.7.0.1 (ref. 55). Quality control was initially performed in Python using Scanpy based on the number of reads (UMI count), number of genes (gene count), percentage of reads mapping to the mitochondrial genome and cell complexity (Supplementary Table 1). Cells with a complexity score (ratio of number of transcripts by genes logged) below 0.7 or more than 30% reads mapping to the mitochondrial genome were removed62,63,64,65,66,67. The mitochondrial threshold was chosen on the basis of published scRNA-seq studies of Anopheles haemocytes62 and Aedes midguts63, noting that a similar threshold (25%) was used by two scRNA-seq studies on the midgut of Drosophila melanogaster64 and Culex tarsalis65, whereas snRNA studies used lower cutoffs due to inherent differences in the technology66,67. The resulting count matrix from each sample was merged into a single AnnData object by Scanpy, before conversion to Seurat using SeuratDisk’s function Convert. Doublet removal was performed in R using scDblFinder68. Quality metrics indicate that mitochondrial percentage is lower in 36 h and 2 dpi samples compared to the 4 dpi and 7 dpi ones, probably owing to a combination of biological factors, including time after blood feeding, age and infectious stages (ookinetes or young oocysts versus large oocysts).
After quality control, mosquito cells from each time point were processed using a similar workflow as for parasite analysis. The top 25% variable genes were identified, the data were scaled, and PCA was performed computing the first 100 PCs. Integration was conducted using RPCA with the highest k.weight (64) to correct batch effect while avoiding overcorrecting for biological variation between time points and treatments. The UMAP was calculated using the first 56 PCs, and cluster identification was based on the Louvain algorithm with a resolution of 0.3. Positive markers were identified using the FindAllMarkers function with a log2 fold change greater than 1. Proventriculus, anterior and posterior midgut scores were calculated based on A. gambiae midgut bulk RNA-seq results69. Clusters were then annotated by identifying the functional enrichment of each cluster’s marker genes as well as cross-referencing cluster markers from four references: the Fly Cell Atlas, the D. melanogaster gut single-cell study, the Aedes aegypti gut single-cell study, and the haemocyte single-cell study from A. gambiae62,64,66,67. A. gambiae orthologues were identified using VectorBase54, and all significant cluster markers with an orthologue specific to a cell type were used for cluster annotation.
Pseudobulk analyses were performed to assess the effect of either EcR knockdown or time post-infectious blood meal on the midgut transcriptome using AggregateExpression from the Seurat package. To assess the effect of EcR knockdown on transcription, differential expression analyses were performed using DESeq2 (ref. 58) by pooling cells from each cell type at each time point. To evaluate the effect of time on transcription, only cells from dsGFP mosquitoes were used to compare between consecutive time points. Functional enrichment analyses were performed using GoProfiler59 when more than 20 genes were significant. Dual-read differential expression analyses of protein coding genes were performed using logistic regression framework comparing dual-read cells at 36 hpi with cells not interacting with a parasite at the same time point, binning for dsRNA treatment and annotation. The analysis was repeated by comparing protein coding gene expression between dual-read ISC/EB cells at 36 hpi and ISC/EBs not interacting with a parasite.
Post-infection exposure assays via sugar feeding
Candidate target selection for drug exposure was based on two criteria: (1) belonging to a distinct gene cluster in our gene model; and (2) having known potent activity against the candidate target in the P. falciparum blood stage. Four drugs were selected: cipargamin (ChemPartner), MMV670325, MMV643121 and MMV007839 (refs. 70,71,72,73) (Medicine for Malaria Venture). Stock solutions were made in DMSO at 100 mM, stored at −20 °C and diluted 1:1,000 in 10% glucose solution shortly before sugar change. As the bioavailability of these compounds in mosquitoes is unknown, a single high concentration of 100 µM was chosen initially as it is close to the solubility limit while keeping DMSO content to 0.1%. Mosquito compound exposure through sugar feeding was performed similarly to previously described53. In brief, sugar solutions containing compounds or corresponding controls (0.1% DMSO) were randomly assigned to a mosquito group and replaced every day from 2 dpi to 10 dpi (unless specified). For oocyst counts, midguts were collected at 7 dpi and stained in 0.2% (w/v) mercurochrome (Sigma-Aldrich). Midguts were imaged using an Olympus Inverted CKX41 microscope, and oocyst number and mean size were calculated using OocystMeter74. For salivary gland sporozoite quantification, salivary glands of individual female mosquitoes were dissected at 14 dpi in PBS and counted on a haemocytometer.
Plasmid construction
The PfSIP2 homology-directed repair (HDR) plasmid was assembled by Golden Gate cloning (New England Biolabs) using purified PCR products of (1) a C-terminal homology region (Cterm-HR) that was codon-altered to avoid repeated CRISPR cuts; (2) a cassette containing haemagglutinin (HA) tags, tet-aptamers75, a TetR-DOZI fusion protein75,76, and a human dihydrofolate reductase (hDHFR) selection marker; (3) a 3′ homology region; and (4) a bacterial backbone. Primers used to amplify fragments are listed in Supplementary Table 8 and assembly was confirmed by whole plasmid sequencing (Plasmidsaurus). Two CRISPR–Cas9 guide plasmids were constructed to cut the C terminus of the endogenous PfSIP2. Guide oligonucleotides were annealed and ligated into BpiI-digested pRR216 (ref. 77), which contains SpCas9, a U6 guide cassette, and a yeast dihydroorotate dehydrogenase (DHODH) selection marker. Sequences were confirmed by Sanger sequencing (Psomagen).
Generation of PfSIP2-cKD parasite line
The HDR and guide plasmids of PfSIP2 were extracted using a Qiagen Maxi kit, and 100 μg of the HDR plasmid was linearized with XhoI, NotI, and ApaL1 at 37 °C overnight. The linearized HDR plasmid and a mixture of the 2 guide plasmids (60 μg each) were sterilized by ethanol precipitation and resuspend in TE buffer. The plasmids were then mixed with 270 µl of Cytomix (120 mM KCl, 0.15 mM CaCl2, 2 mM EGTA, 5 mM MgCl2, 10 mM K2HPO4, 10 mM KH2PO4, 25 mM HEPES, pH 7.6) and transfection by red blood cell loading was performed as described78. The parasite line was maintained at 5% haematocrit with 500 nM ATC (Sigma) from the onset of transfection. Drug pressure for the guide plasmid was applied from 6 h to 5 days post-transfection using 1.5 μM DSM1 (Sigma), whereas drug pressure for the HDR plasmid was maintained with 2.5 nM WR99210 hydrochloride (Sigma) starting 6 h post-transfection until integration was confirmed. To do so, genomic DNA was extracted from WT and PfSIP2-cKD lines using the Qiagen Blood & Tissue kit. Each integration site of the double crossover was amplified as well as the whole locus and PCR products were analysed by gel electrophoresis.
Asexual stage growth assay
ATC was washed out from PfSIP2-cKD parasites three times with RPMI 1640. The resulting parasites were diluted to 0.5% starting parasitaemia at 2.5% haematocrit and treated with either 500 nM ATC, 1.5 nM ATC or 0.025% of DMSO. Parasitaemia was calculated daily by counting Giemsa-stained smears of each technical replicate. The experiment was repeated twice, with four technical replicates for each biological replicate. Data were analysed by two-way ANOVA with Dunn’s correction.
Mosquito infection with transgenic parasites
Transgenic parasites were cultured in the asexual blood and gametocyte stages and fed to mosquitoes as described above, with the following specifications for ATC usage. Both asexual and gametocyte cultures were maintained in 500 nM ATC (dissolved in DMSO). One hour prior to mosquito infection, ATC was washed out from the gametocyte cultures by replacing the media three times, and either 500 nM ATC or 0.025% DMSO was added to the blood meal. Mosquitoes were maintained on a 10% glucose solution supplemented with 100 µM ATC, which was dissolved directly in the solution.
Culturing of primary human hepatocytes and infection with transgenic parasites
Cryopreserved human primary hepatocytes (BioIVT) were thawed, seeded on collagen-coated micropatterned islands in 96 well plates, and infected with WT or PfSIP2-cKD sporozoites as previously described79. In short, WT or PfSIP2-cKD-infected mosquito salivary glands were dissected in Schneider’s medium (Gibco) containing 200 U ml−1 of penicillin, 200 µg ml−1 of streptomycin (2×, Gibco) for less than 1 h. Sporozoites were released from glands, filtered through a 35 µm cell strainer and counted on a haemocytometer. Parasites were spun at 10,000g for 3 min, resuspended in hepatocyte media containing 2.5 μg ml−1 Fungizone (Cytiva) and 2× penicillin/streptomycin as well as either 500 nM ATC or 0.025% of DMSO and seeded onto hepatocytes. For invasion assays, 7,000 sporozoites were seeded per well, wells were fixed in 4% paraformaldehyde 3 hpi and in and out PfCSP staining was performed79. For infection assays, 70,000–100,000 sporozoites were used, and subsequently washed off 3 hpi before supporting cells from the male mouse fibroblast cell line 3T3-J2 (ref. 80) were added. Cells were washed daily and wells were fixed at 2 dpi in 100% methanol and stained with rabbit PfHSP70 monoclonal primary antibody (antibodies-online, ABIN361730) and goat anti-rabbit IgG 546.
Immunofluorescent microscopy
Midguts were dissected from female mosquitoes at specified time points. Any remaining blood bolus was removed in ice-cold PBS when present and clean midguts were incubated at room temperature in 4% paraformaldehyde for 45 min, followed by 3 washes in PBS for 10 min each. Midguts were permeabilized and blocked in 0.1% Triton X-100, 3% bovine serum albumin (BSA) in PBS for 1 h at room temperature or overnight at 4 °C before incubation in primary antibody at room temperature for 2 h, shaking. The following primary antibodies were used: 1:200 anti-Toxoplasma gondii centrin-1 rabbit polyclonal (Kerafast EBC004); 1:5 anti-Drosophila armadillo mouse monoclonal supernatant N2 7A1 (ref. 81) (deposited to the DSHB by E. Weischaus), 1:200 anti-Pfs25 mouse monoclonal 4B7 (ref. 82) (deposited to BEI resources by L. H. Miller and A. Saul). Another 3 washes in PBS were performed before incubating the samples in secondary antibodies and phalloidin for 1 h at room temperature rocking (goat anti-rabbit IgG AlexaFluor 488, goat anti-mouse IgG AlexaFluor 488, goat anti-mouse AlexaFluor 568 from Invitrogen used at 1:400, Ebioscience Phalloidin eFluor 660 used at 1:300). Midguts were then stained in PBS containing 5 µg ml−1 DAPI before the remaining 3 washes in PBS only. Midguts were mounted using Vectashield HardSet Antifade Mounting Medium and imaged on a Zeiss Inverted Observer Z1 with Apotome3 or a Zeiss LSM 980 confocal microscope. Using ZEN 3.10 software, confocal images were processed with LSM Plus whereas Airyscan images were processed with default parameters, followed by brightness and contrast adjustments in Fiji (v.2.14.0). Gamma was not adjusted unless specified.
Statistical analysis
Specific statistical tests are detailed in the corresponding figure legend. Confirmation of EcR knockdown and analysis of parasite prevalence, intensity and infection rates were conducted using GraphPad Prism 10.1.1. Odds ratios of dual-read parasites in mosquito cell types were calculated in R using Fisher’s exact tests followed by correction for multiple comparisons with Benjamini–Hochberg multiple comparison correction.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
First Appeared on
Source link

