Parasites trigger epithelial cell crosstalk to drive gut–brain signalling
Mice
All experimental procedures were conducted in accordance with guidelines approved by the Institutional Animal Care Committees at the University of California, San Francisco (UCSF) and the South Australian Health and Medical Research Institute (SAHMRI) and aligned with the NIH and NHMRC Guide for the Care and Use of Laboratory Animals, respectively. Mice of both sexes, aged 8–16 weeks, were used, and given ad libitum access to standard lab chow and sterile water. They were housed in a controlled environment under a 12-h light–dark cycle. TRPM5-GFP mice were used to visualize tuft cells (gift from R. F. Margolskee). Pou2f3−/− mice (Jackson Laboratory, strain 037040) were used to knock out tuft cells. For conditional knockout of Chat in intestinal tuft cells, VilCre mice (Jackson Laboratory, strain 021504) were crossed to Chatflox mice (gift from J. Chan). For gGRAB5-HT3.0 sensor imaging, VilCre mice were crossed to the Rosa26gGRAB-5-HT3.0-P2A-jRGECO1a reporter line. GCaMP imaging in organoids used Tac1Cre mice (Jackson Laboratory, strain 021877) crossed with Polr2aGCaMP5g-IRES-tdTomato mice (gift from L. Jan, Jackson Laboratory, strain 024477). To inhibit serotonin release from EC cells, we expressed the tetanus toxin in EC cells using the Cre- and Flp-dependent tetanus toxin light-chain reporter mouse, RC::PFTox (gift from S. Dymecki). Specifically, ePetFlp hemizygous/RC::PFTox homozygous mice were crossed to Tac1Cre homozygous mice to produce Tac1Cre;ePetFlp;PFTox mice. For nerve fibre recordings, Scn10aCre mice (gift from W. Imlach, Jackson Laboratory, strain 036564) were crossed to the ChR2 reporter line (Rosa26lsl-ChR2; Jackson Laboratory, strain 012569). Genetically modified mice or control mice (littermates or age-matched mice) were randomly selected for all behavioural experiments.
Crypt cell isolation and organoid culture
Adult male Tac1Cre;Polr2aGCaMP5g-IRES-tdTomato, TRPM5-GFP and Pou2f3−/− mice were used to generate intestinal organoids as previously reported51. Approximately 8-cm pieces of the ileum were used to establish TRPM5-GFP organoids. For Tac1Cre;Polr2aGCaMP5g-IRES-tdTomato organoids, the upper jejunum was used to avoid ectopic expression of Tac1Cre in the lower intestine. Organoids were maintained and passaged every six days in organoid growth medium (advanced Dulbecco’s modified Eagle’s medium (DMEM)/F12 supplemented with penicillin–streptomycin, 10 mM HEPES, GlutaMAX, B27 (Thermo Fisher Scientific), 1 mM N-acetylcysteine (Sigma), 50 ng ml−1 mouse recombinant epidermal growth factor (Thermo Fisher Scientific), R-spondin 1 (10% final volume) and 100 ng ml−1 mouse Noggin (Peprotech)).
Cell lines
The R-spondin-1-expressing HEK293FT (ATCC) cells were maintained in DMEM, 20% fetal calf serum (FCS), 1% penicillin–streptomycin and 125 µg ml−1 zeocin (Thermo Fisher Scientific) at 37 °C, 5% CO2. Zeocin was removed after production of R-spondin 1 conditioned medium. HEK293T cells (ATCC) were grown in DMEM, 10% FCS and 1% penicillin–streptomycin at 37 °C, 5% CO2 and transfected using Lipofectamine 3000 (Thermo Fisher Scientific) according to the manufacturer’s protocol. For biosensor experiments, 200 ng pDisplay-gGRABACh4h-IRES-mCherryCAAX or 200 ng pcDNA3-hM1R-P2A-GCaMP8m was transfected to HEK293FT cells in 24-well plates. For the 5-HT3 biosensor experiment, 200 ng pcDNA3-5-HT3A and 20 ng pcDNA3-mApple were co-transfected to HEK293FT cells in 24-well plates.
Induction of tuft cell hyperplasia
Tuft cell hyperplasia in organoids was induced by exposing organoids to 20 ng ml−1 IL-4 (R&D Systems) in the growth medium for two days (days 3–5), followed by one day of growth in IL-4-free medium. The biosensor experiments were then performed on day 6. To induce tuft cell hyperplasia in mice, the mice received intraperitoneal injections of 500 ng of IL-25 (R&D Systems) on days 0, 1, 2 and 3. Tissues were subsequently collected for imaging on day 5.
GCaMP imaging using intestinal organoids
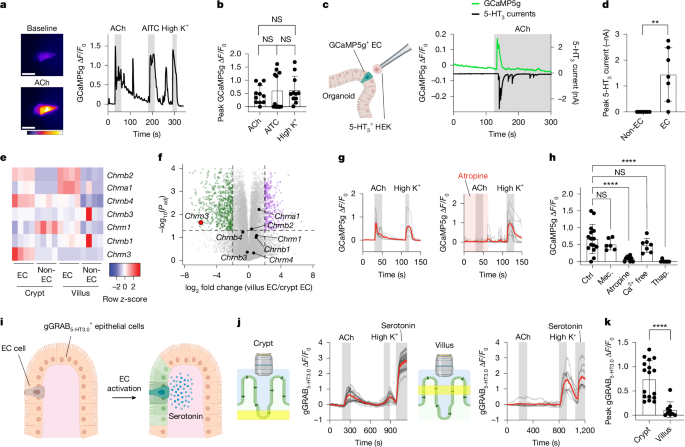
Five days after passage, Tac1Cre;Polr2aGCaMP5g-IRES-tdTomato organoids were removed from Matrigel (Corning) and mechanically broken up with a 200-µl pipette. The organoid fragments were seeded onto Cell-Tak (Corning)-coated coverslips and placed in a recording chamber containing Ringer’s solution (140 mM NaCl, 5 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 10 mM d-glucose and 10 mM HEPES-Na (pH 7.4)). EC cells were identified by tdTomato expression. GCaMP imaging was performed with an upright microscope equipped with a Grasshopper 3 (FLIR) camera and a Lambda LS light source (Sutter). Organoids were maintained under a constant laminar flow of Ringer’s solution applied by a pressure-driven microperfusion system (SmartSquirt, Automate Scientific). All pharmacological reagents were delivered by local perfusion. Acquired images were analysed with Fiji. Regions of interest (ROIs) were drawn around individual EC cells, and ∆F/F0 was calculated.
M1R–GCaMP8m biosensor experiments
Tuft cell hyperplasia was induced in organoids as described above. In each well of 24-well plates, the organoid medium was replaced with 1 ml of Ringer’s solution and incubated for 10 min to remove residual growth medium. Subsequently, the Ringer’s solution was replaced with 300 µl fresh Ringer’s solution and incubated for two hours, after which the supernatant was collected for imaging. For the preparation of M1R–GCaMP8m biosensor cells, HEK293FT cells transiently transfected with pcDNA3-M1R-P2A-GCaMP8m were dissociated with trypsin and plated onto 5-mm glass coverslips. After one hour, coverslips were moved to µ-Slide III 3D Perfusion chamber slides (ibidi). Imaging was performed using an upright microscope equipped with a Grasshopper 3 camera (FLIR) and a Lambda LS light source (Sutter Instrument). The entire area of each biosensor cell was used for the calculation of ∆F/F0 values. The organoid supernatant was manually applied with a 200-µl pipette. All images were analysed using Fiji software v.2.14 (NIH).
gGRABACh4h biosensor experiments
HEK293FT cells transiently transfected with pDisplay-gGRABACh4h-IRES-mCherryCAAX were dissociated with trypsin and washed once with Ringer’s. The dissociated cells were plated on top of intestinal organoids. For biosensor experiments with isolated tuft cells and TRPM5-GFP organoids, individual HEK293FT cells were carefully lifted from coverslips and positioned 5 µm from a tuft cell using a glass pipette. In other gGRABACh4h biosensor experiments, signals were measured from biosensor cells surrounding the organoids. Imaging was performed using an upright microscope equipped with a Grasshopper 3 camera (FLIR) and a Lambda LS light source (Sutter Instrument). The entire area of each biosensor cell was used for the calculation of ∆F/F0 values. The bath solution was static to prevent the washout of endogenously released ACh, and 10 µM TRPM5 agonist 39 (ProbeChem), 10 µM NDNA (ProbeChem) and 10 mM succinate were applied manually with a 1,000-µl pipette. All images were analysed using Fiji.
5-HT3 biosensor experiments
HEK293FT cells transiently transfected with pcDNA3-5-HT3A and pcDNA3-mApple were dissociated with trypsin and washed once with Ringer’s. The dissociated cells were plated on top of intestinal organoids. Transfected cells were identified based on the mApple expression, and whole-cell configuration was achieved. While the membrane potential was held at −80 mV, individual HEK293FT cells were carefully lifted from coverslips and positioned 5 µm from an EC cell using a glass pipette. GCaMP imaging of organoid EC cells was performed using an inverted microscope equipped with a Grasshopper 3 camera (FLIR) and a Lambda LS light source (Sutter Instrument). The bath solution was static to prevent the washout of endogenously released serotonin, and pharmacological agents were applied manually with a 1,000-µl pipette. All images were analysed using Fiji.
Patch-clamp recordings
Recording was performed with pClamp software v.10.7 (Molecular Devices) using a Multiclamp 700A amplifier (Molecular Devices) connected to a Digidata 1550B digitizer (Molecular Devices). Patch electrodes (2–4 MΩ) were pulled from borosilicate capillaries (BF-150-110-10, Sutter Instrument). The intracellular solution consists of 140 mM K-aspartate, 13.5 mM NaCl, 1.6 mM MgCl2, 0.09 mM EGTA and 9 mM HEPES-K (pH 7.35). Recordings were performed in Ringer’s solution. For measuring voltage-gated calcium currents, the following solutions were used: (external) 140 mM NMDG, 10 mM BaCl2, 10 mM HEPES and 12.5 mM glucose (pH 7.4); (internal) 140 mM CsMeSO4, 2 mM MgCl2, 0.1 mM CaCl2, 10 mM HEPES and 5 mM EGTA (pH 7.4). TRPM5 agonist 39 (2 µM; ProbeChem) and 10 mM succinate reagents were delivered locally by gravity-driven microperfusion (Valvebank, Automate Scientific).
Ex vivo serotonin sensor imaging
An approximately 1 cm piece of the jejunum was isolated from an 8–16-week-old VilCre;Rosa26gGRAB-5-HT3.0-P2A-jRGECO1a mouse. For recordings from VilCre;Rosa26gGRAB-5-HT3.0-P2A-jRGECO1a;Pou2f3−/− or VilCre;Rosa26gGRAB-5-HT3.0-P2A-jRGECO1a;Chatf/f mice, genetically modified mice or control mice (littermates or age-matched mice) were randomly selected. The tissue was immediately filleted open along the mesenteric border, and the smooth-muscle layer was manually removed for tissue stability. Tissue segments were pinned down to a Sylgard-coated recording chamber, and imaged from the smooth-muscle side to observe crypts and from the luminal side to observe villi. Imaging was performed with a Leica SP8 confocal microscope with an HC APO L 20×/1,00 W objective and LAS X software (Leica Microsystems). The tissue was bath-perfused with bubbled room-temperature Krebs buffer (118 mM NaCl, 4.7 mM KCl, 2 mM MgCl2, 2 mM CaCl2, 1.2 mM KH2PO4, 25 mM NaHCO3 and 10 mM d-glucose) at a rate of around 1 ml per min. All pharmacological reagents were diluted in Krebs buffer and bath-perfused with simultaneous manual application. For measuring baseline serotonin levels, the gGRAB5-HT3.0 sensor was fully activated by bath-applied 20 µM serotonin, followed by the complete quenching of gGRAB5-HT3.0 with 50 µM RS 23597-190 (Tocris), a 5-HT4 receptor antagonist. For baseline serotonin measurements after Nb infection, mice were infected subcutaneously with 500 third-stage Nb larvae (L3) and euthanized on day 9 for imaging. Acquired images were analysed with Fiji. ROIs were drawn around individual crypts or villi and ∆F/F0 was calculated and normalized to serotonin-activated maximum signals. AUC was calculated as ∑(Normalized gGRAB5-HT3.0 ∆F/F0) for the duration of 3 min during baseline.
Dissociation of tuft cells
The ileum from TRPM5-GFP mice was cut into approximately 3-cm segments, incubated in 10 ml of cold Dulbecco’s phosphate-buffered saline (DPBS) with 2 mM EDTA on ice for 12 min, then transferred to 6 ml of warm DPBS with 2 mM EDTA and incubated at 37 °C for 8 min. To dissociate the epithelial layer, vigorous shaking was applied for 30–60 s. The dissociated epithelium was centrifuged and washed with DPBS containing 10% fetal bovine serum (FBS). The washed epithelium was digested in 10 ml digestion buffer (Hanks’ balanced salt solution (HBSS) with 0.2 mg ml−1 dispase II (Sigma) and 0.2 mg ml−1 DNaseI (Sigma)) at 37 °C for 4 min, with vigorous shaking at 2-min intervals. The cells were then washed once with HBSS containing 10% FBS and 0.2 mg ml−1 DNaseI, filtered through 70-µm and 40-µm strainers and resuspended in cold Ringer’s solution supplemented with 5 µM Y-27632 (Sigma). Cells were kept on ice and used within two hours.
Dissociation of EC cells
For GCaMP imaging, EC cells were isolated from the upper half of the small intestine of 8–16-week-old Tac1Cre;Polr2aGCaMP5g-IRES-tdTomato mice. To isolate crypt and villus epithelial cells, intestinal tissue was cut into approximately 3-cm segments and filleted, and the villus epithelium was dissociated by gentle scraping with a glass coverslip. The remaining tissue was subsequently incubated in 10 ml of cold DPBS with 30 mM EDTA and 1.5 mM dithiothreitol (DTT) on ice for 20 min. The tissue was transferred to 6 ml of pre-warmed DPBS with 30 mM EDTA and incubated at 37 °C for 8 min, followed by 30–60 s of vigorous shaking to dissociate the crypt epithelium. The dissociated crypt and villi fractions were washed with 10 ml DPBS with 10% FBS and digested in 10 ml HBSS with 0.6 mg ml−1 dispase II [Sigma] and 0.2 mg ml−1 DNaseI (Sigma)) at 37 °C for 12 min, with vigorous shaking at 2-min intervals. The cells were then washed with 10 ml HBSS with 10% FBS and 0.2 mg ml−1 DNaseI. For GCaMP imaging, dissociated crypt cells were filtered through 70-µm and 40-µm strainers, resuspended in DMEM supplemented with 10% FBS, B27 and 5 µM Y-27632 (Sigma) and plated onto glass coverslips precoated with 2.5% Matrigel solution. Three days after dissociation, healthy EC cells were identified by tdTomato fluorescence and characteristic polygonal or cone-shaped morphology.
Brainstem tissue collection
Mice received an intraperitoneal injection of 500 ng IL-25 or saline every 24 h for a duration of 4 days. To minimize background FOS signals in the nTS region, mice were fasted overnight from day 3. On day 4, four hours after receiving the final injection, mice were euthanized for tissue collection. For helminth infection experiments, mice were euthanized for tissue collection 11 days after infection. Whole-body perfusion was performed for each mouse with 30 ml ice-cold DPBS (Thermo Fisher Scientific) followed by 20 ml ice-cold 4% paraformaldehyde (PFA). The whole brain and brainstem were then collected and post-fixed overnight at 4 °C in 4% PFA. After fixation, brainstems were dissected and washed thoroughly in PBS, followed by 30% sucrose dehydration at 4 °C overnight. The brainstems were then cryo-embedded in OCT (Sakura) for cryosections. Ten-micrometre sections were collected from Bregma −6.92 mm to −8.00 mm to cover the whole nTS region that receives extensive projection from the gut-innervating vagal afferents30.
Histology and immunostaining
Immunofluorescence in the intestine and brainstem was performed using 10-µm cryosections. For staining organoids, whole organoids were fixed in 4% PFA for 30 min at room temperature. Immunofluorescence was then performed with whole organoids or 7-µm cryosections. Blocking was performed with 5% w/v bovine serum albumin (BSA; Sigma), 5% normal serum corresponding to secondary antibody species and 0.3% Triton-X in PBS at room temperature for 30 min. Primary antibodies were incubated overnight at 4 °C at the indicated dilutions. The antibodies used were against DCAMKL1 (1:250, Abcam), GFP (1:300, Abcam) and FOS (1:300, Synaptic Systems). Secondary antibodies from Invitrogen (Alexa Fluor 647 goat anti-rabbit, Alexa Fluor 647 goat anti-rat, Alexa Fluor 568 goat anti-rabbit, Alexa Fluor 568 donkey anti-rat, Alexa Fluor 488 goat anti-chicken and Alexa Fluor 488 donkey anti-chicken) were incubated at a 1:500 dilution for two hours at room temperature at a 1:500 dilution. All the imaging was performed at the UCSF Center for Advanced Light Microscopy. Confocal images were captured on an inverted Nikon Ti microscope run using Micro Manager 2.0 Gamma52, equipped with a Zyla 4.2 CMOS camera (Andor), piezo XYZ stage (ASI), CSU-W1 spinning disk with Borealis upgrade (Yokogowa/Andor), Spectra-X (Lumencor) and ILE 4 line Laser Launch (405/488/561/640 nm; Andor). Images were taken using a Plan Apo λ 20×/0.75 and Plan Apo VC 60×/1.4 Oil using lasers 405, 488, 561 and 647 nm and emission filters 447/60, 525/50, 607/36 and 645/65 for DAPI, GFP, RFP and Cy5, respectively. Large, stitched images were taken using the same set-up if necessary. Maximum-intensity projections were generated in Fiji.
In situ hybridization
Cryosections (5 or 10 μm) were prepared as described above. RNA fluorescence in situ hybridization (RNA-FISH) was performed using a RNAscope Multiplex Fluorescent Detection Kit v.2 (Advanced Cell Diagnostics) according to the manufacturer’s protocol. The following probes were used in this study: Mm-Dclk1-C2 (476631-C2), Mm-Chat (408731), Mm-Sucnr1 (437721), Mm-Fos-C2 (316921-C2), Mm-Npy (313321), Mm-Dbh (407851) and Mm-Nr4a2 (423351). Z-stack images were taken with a Nikon CSU-W1 spinning disk confocal microscope as described above (UCSF Center for Advanced Light Microscopy). Maximum-intensity projections were generated using Fiji (v.2.14).
Brainstem FOS quantification
We selected the medial part of gut-innervating vagal afferents that project to the nTS (−7.32 mm to −7.76 mm) region for the quantification of activated neurons, because we found few FOS+ signals in the rostral (Bregma −6.92 mm to −7.32 mm) or caudal (Bregma −7.76 mm to −8.00 mm) parts. Each data point represents one mouse, and the numbers of FOS+ neurons were averaged across multiple slides per individual. At least 15 (PFTox study) or 8 (colocalization study and helminth study) slides from each individual were analysed. For colocalization analysis, we performed IL-25 injections on Tac1creRosa26GCaMP5g-IRES-tdTomato mice and stained GFP to visualize Tac1+ neurons. In IL-25–injected wild-type mice, we performed RNAscope co-staining of Fos with Dbh, Npy or Nr4a2 to visualize Dbh+, Npy+ and Nr4a2+ neurons, respectively. CCK8 staining was used to visualize Cck+ neurons. Comparable numbers of males and females were used in each study, with no sex differences detected.
Food intake assay
For food intake measurements with IL-25 injections, 8–12-week-old mice were acclimated to metabolic cages (41700UB, Animalab) for 11 days. Genetically modified mice or control mice (littermates or age-matched mice) were randomly selected. A standard laboratory powder diet (5053, PicoLab Rodent Diet) was provided. Mice were adjusted to the cage environment for three days. After this adjustment period, body weight and food and water intake were measured every 24 h. After 3 days of baseline measurements, mice received an intraperitoneal injection of either 500 ng IL-25 or saline every 24 h for a duration of 6 days.
To measure food intake after acute tuft cell stimulation, 8–12-week-old mice were singly housed and acclimated to FED3.1 automated feeding devices (OEPS-7510, Open Ephys) for 2 days before experimentation. The FED3.1 system continuously monitored and recorded individual feeding events, providing precise measurements of food consumption. After the acclimation period, a two-day experimental protocol was implemented using a within-subject design. On day 1, mice received oral gavage administration of 200 μl saline as a vehicle control at 18:00, immediately before the onset of the dark cycle. Food intake was continuously monitored overnight for 12 h using the FED3.1 system. On the following day (day 2), the same mice received oral gavage administration of 200 μl of TRPM5 agonist 39 (100 μM dissolved in saline) at 18:00 to stimulate tuft cell activation. Food intake was again monitored overnight for 12 h after treatment.
For food intake measurements during Nb infections, 10–13-week-old mice were singly housed and acclimated to controlled feeding devices (929102, Research Products International) for 2 days before infection. The feeding system was loaded with 20 mg precision pellets (F0071, BioServ) to enable accurate quantification of food consumption throughout the infection period. Infectious third-stage Nb larvae (L3) were raised and maintained as previously described53. On day 0, mice were infected subcutaneously with 500 Nb L3. Food intake was continuously monitored from the day of infection (day 0) to day 11 after infection using the controlled feeding devices. Daily food consumption was recorded as total pellet intake (g).
Ex vivo nerve fibre recordings of mucosal sensory afferents innervating the small intestine
Jejunal recordings were performed in 12–18-week-old male and female wild-type and VilCre;Chatflox/flox mice, as previously described5. In brief, mice were euthanized by CO2 inhalation, and a small section of the jejunum (around 2 cm) was removed along with the attached neurovascular bundle. Jejunum segments were opened longitudinally and pinned flat, mucosal side up, in a specialized organ bath consisting of two adjacent compartments. The jejunal compartment was perfused with Krebs solution (117.9 mM NaCl, 4.7 mM KCl, 25 mM NaHCO3, 1.3 mM NaH2PO4, 1.2 mM MgSO4, 2.5 mM CaCl2 and 11.1 mM d-glucose), bubbled with carbogen (95% O2, 5% CO2) at a temperature of 34 °C. Krebs solution also contained 1 µM nifedipine (to suppress smooth-muscle activity) and 3 µM indomethacin (to suppress potential inhibitory actions of endogenous prostaglandins). The end of the neurovascular bundle, which contains the vagus nerve, was extended from the tissue compartment into the paraffin-oil-filled recording compartment and laid onto a mirror. The whole nerve bundle supplying the jejunal segment was then located within the neurovascular bundle, carefully cleaned away and placed on a platinum recording electrode. Action potentials generated within the jejunum travelled along the nerve fibres, through to the recording electrode and into a differential amplifier. These signals were filtered, sampled (20 kHz) using a 1401 interface (CED) run by Spike 2 software (v.5.18) and stored on a PC for off-line analysis.
Optogenetic stimulation of jejunal mucosal afferents
Scn10aCre;Rosa26lsl-ChR2 mice, which express ChR2 in sensory afferents but not in EC cells, were used4,5. This transgenic line enabled us to optogenetically activate mucosal afferents in an EC-independent and mechanically independent manner, as previously described. Using the jejunal ex vivo afferent preparation described above, we recorded the action potentials generated by stimulating an approximately 3-mm2 section of the jejunum with continuous light (470 nm) at increasing intensities (0.08–7 mW; 2 s exposure each intensity and a 10-s interval between exposures). Light was delivered using a High Power Fiber-Coupled LED Light Source (model BLS-FCS-0470-10) and Multimode Fiber Patchcords (numerical aperture: 0.39 NA; core size: 400 µm; FPC-0400-39-025MA-BP, Mightex). After this graded illumination protocol, we perfused (by gravity) the section of the jejunum receiving the light stimuli with 10 µM ACh, succinate (10 mM) or TRPM5 agonist 39 (10 µM). These agonists were perfused alone or after 10 min perfusion with (and still in the presence of) the 5-HT3 antagonist alosetron (10 μM). Five minutes after perfusion of the compounds, we repeated the graded illumination protocol described above. Action potentials generated by light stimuli were analysed off-line using the Spike 2 wavemark function and discriminated as single units on the basis of a distinguishable waveform, amplitude and duration (CED). Data are expressed as either: (1) afferent activity induced by individual light stimuli (action potentials per s) or the compounds tested; or (2) light intensity threshold for action potential activation (mW mm−2). Data were analysed to determine whether they were normally distributed using Shapiro–Wilk tests, with subsequent analysis using two-way ANOVA with two-tailed Wilcoxon matched-pairs rank test, or Kruskal–Wallis test with Dunn’s multiple comparison (for more than two groups). For comparisons between two groups, two-tailed paired t-tests were used. P < 0.05 was considered statistically significant throughout.
Behavioural assays
To induce tuft cell hyperplasia, wild-type, PFTox(+), Pou2f3−/− and Vilcre;Chatflox/flox mice were injected intraperitoneally daily on four consecutive days with either sterile saline (vehicle) or 500 ng IL-25. In the morning on day 5, mice were transferred from their individually ventilated cages (IVCs) to a temperature-controlled test room (22 ± 1 °C) and allowed to acclimatize for at least 15 min before testing. Assessment of locomotor activity and spontaneous behaviour in mice was evaluated using a behavioural spectrometer (Behavior Sequencer, Behavioral Instruments and BiObserve). This consisted of a 40 × 40-cm square arena enclosed at a height of 45 cm, with an aluminium floor on vibration sensors and a ceiling centre-mounted camera. In addition, the spectrometer is equipped with a row of 32 infrared transmitters and receiver pairs embedded in the walls at a height of 6.5 cm and halogen strip lights illuminating the inside of the behavioural box.
For testing, two main groups of mice were studied: (1) mice orally gavaged with vehicle (200 µl saline) or the TRPM5 agonist (TRPM5 agonist 39, 200 µl bolus at 100 µM); or (2) mice intraperitoneally administered vehicle or IL-25. Mice were individually placed in the centre of the behavioural spectrometer and their behaviours were filmed, tracked and analysed by computerized video tracking software (Viewer, BiObserve) for a total duration of 20 min. Total distance travelled in the open field (cm), time spent in the central area (20 × 20 cm) (s), wall distance (cm) and time spent (s) in 23 different behavioural patterns, including grooming, orienting, rearing and forms of locomotion, were evaluated. All recording sessions were performed between 09:30 and 12:30 each day in an order counterbalanced for the experimental group (maximum of six mice per day). Experimenters were blinded for all behavioural tests.
Abdominal electronic von Frey Hair testing
Mechanical allodynia was assessed in vivo using electronic Von Frey hairs (eVFH) in C57BL/6 (Jackson Laboratory) mice that were intraperitoneally injected with vehicle or IL-25. Abdominal withdrawal thresholds caused by gradually applied mechanical pressure to the lower abdomen were measured as previously described54. Before eVFH testing, on days 2 and 3 of intraperitoneal IL-25 and vehicle treatment, mice were habituated to the eVFH testing enclosure (clear plexiglas observation chamber, around 230 × 240 × 146 mm, divided into 6 compartments; BSBIOPVF, Panlab), mounted on an elevated wire mesh stand, for around 30 min each day. Habituation was designed to reduce the natural explorative behaviour animals display when they are introduced to a new environment. Therefore, on the eVFH testing day, mice will explore less, which facilitates probing the abdominal area with the eVFH filament.
On day 5, mice were individually placed into the eVFH testing enclosure mounted on the raised grid stand and allowed to acclimatize for a minimum of 15 min. Once mice were still and quiet, abdominal withdrawal thresholds were tested using a portable force transducer equipped with a spring tip (BSBIOEVF4S, Panlab) recording device with corresponding software (BIOCIS Force Ramp software; for automatic recording of results on a PC via an RS232/USB port), and a remote foot switch. The eVFH spring tip, mounted on the handheld force transducer, was gently lifted from below to the lower abdomen of the mouse, and force was gradually applied until the abdomen was withdrawn. The maximum force applied (in grams; with a resolution of 0.1 g) that elicited the abdominal withdrawal was recorded as the individual withdrawal threshold value. Abdominal withdrawal thresholds were measured in the same mouse five times, and the results were expressed for each mouse as a mean abdominal withdrawal threshold. All measurements were performed by the same investigator to ensure consistency. At the end of testing, mice were placed in their home cage and returned to the IVC rack in the testing room to recover for the subsequent assessment of nest building, which was performed as an overnight test (as described below). Experimenters were blinded for all tests.
Assessment of nest building
Nesting behaviour is a natural activity in rodents, which is important for providing shelter and heat conservation. This animal instinct can be used to assess wellbeing, because animals in discomfort will be less inclined to build nests or maintain them55,56. Nesting behaviour was assessed in vivo in mice treated intraperitoneally with either vehicle or IL-25 for five consecutive days to induce tuft cell hyperplasia. In brief, mice were kept in their home cage (individually housed from previous testing) with their original wood chip bedding and food, but all environmental enrichment items (tunnels, tissues and existing nesting material) were removed. A fresh, pre-weighed nestlet, consisting of an approximately 5-mm-thick pressed cotton square (50 mm × 50 mm, Able Scientific, Australia) weighing about 2.5–3 g, was placed in the cage and mice were left undisturbed overnight from 16:00 until 09:00 the following day. In the morning of day 6, the nest quality was assessed manually and scored according to a five-point nest-rating scale protocol55. Score 1: nestlet not noticeably touched (more than 90% intact); score 2: nestlet partially torn (50–90% remaining intact); score 3: nestlet mostly shredded, with material broadly spread in cage without clear nest area (less than 50–10% remaining intact); score 4: nestlet more than 90% torn and a nest area is present in a corner of the cage (nest is flat with less than 50% of the circumference built up higher than the mouse’s body); score 5: more than 90% of the nestlet is torn and the nest has a clear crater with high fluffy walls over more than 50% of the circumference. If the score fell between two scores (for example, scores 3 and 4), the score was split, and a mean value was noted (for example, 3.5). In addition to evaluating the quality of the nest, any unshredded nestlet material left (intact nestlet pieces weighing more than 0.1 g) after a bout of nesting was weighed, and the percentage of nestlet material shredded (% nestlet shredded) was determined for each mouse. Experimenters were blinded for all tests.
Retrograde tracing of mucosal afferents from the small intestine
Adult male C57BL/6 mice (Jackson Labs) aged 14–19 weeks were used for vagal ganglia collection. Mice were housed in groups (two to five mice per cage) in a specific and opportunistic pathogen-free facility, fed a Jackson lab diet, provided with environmental enrichment (shelter, nesting material and so on) and had normal immune status. Retrograde tracing using cholera toxin subunit B (CTB, 0.5%) directly conjugated to Alexa Fluor AF594 (for calcium imaging) or Alexa Fluor AF488 (for PCR with reverse transcription; RT–PCR) (C234777 and C22841, Invitrogen, Thermo Fisher Scientific) was performed from the lumen of the proximal small intestine. A small aseptic abdominal incision was made in mice anaesthetized with isoflurane (2–4% in 0.6 l min−1 oxygen). The proximal small intestine was located, and 5-μl injections were made through the intestinal wall into the lumen at three to four sites covering 5 cm of the proximal small intestine. The tracer was expelled completely before the withdrawal of the needle back through the intestine wall. Injections were made with a 30-gauge needle (HAMC7803-07, point style: 4 (10–12); Hamilton Company, Bio-Strategy) attached to a Hamilton 5-μl syringe (HAMC7634-01 5 µl 700 series RN syringe; Hamilton Company, Bio-Strategy). The intestinal walls were gently rubbed together using cotton tip applicators to distribute the tracer throughout the lumen and the abdominal incision was sutured closed. Analgesic (buprenorphine, 0.1 mg per kg) and antibiotic (ampicillin, 50 mg per kg) were administered via subcutaneous injection as mice regained consciousness. Mice were then housed individually and closely monitored for four days before vagal ganglia collection for downstream cell picking for single-cell RT–PCR or Ca2+ imaging studies.
Cell culture for Ca2+ imaging and single-cell RT–PCR of small-intestine mucosal-traced vagal neurons
After mucosal tracing, vagal ganglia were isolated from three male C57Bl/6J mice and enzymatically dissociated as previously described5. Ganglia were placed in 3 ml of magnesium- and calcium-free HBSS (Gibco) with 12 mg of collagenase II (Gibco, Thermo Fisher Scientific) and 14 mg of dispase (Gibco) for 30 min at 37 °C. The enzyme solution was then replaced with 3 ml HBSS containing 12 mg collagenase II for an additional 10 min at 37 °C. Ganglia were washed twice with HBSS and then triturated through a fire-polished Pasteur pipette until a single-cell suspension was achieved. The cell suspension was centrifuged at 50g for one minute and the cell pellet was resuspended in DMEM (25 mM glucose, 1 mM pyruvate; Gibco) containing 10% FCS (Invitrogen), 1× GlutaMAX (Gibco), 1× MEM non-essential amino acids (Gibco), 1× penicillin–streptomycin (Invitrogen) and 100 ng ml−1 NGF (Merck). Neurons were spot-plated on coverslips coated with poly-d-lysine (Merck, 100 µg ml−1) and laminin (Merck, 18 µg ml−1) and maintained at 37 °C in 5% CO2.
Single-cell RT–PCR of mucosal-traced vagal ganglion neurons
CTB-traced neurons were manually picked using a micromanipulator under a fluorescent microscope. Cells were under a continuous slow flow of RNA/DNase-free PBS to reduce potential contamination. After picking a traced cell, the glass capillary was broken into a tube containing 9 μl lysis buffer with 1 μl DNaseI (Single Cell-to-CT qRT-PCR kit; Thermo Fisher Scientific) and further processed according to the manufacturer’s instructions. A bath control was taken and analysed from every coverslip along with the other samples. PCR was performed according to the manufacturer’s instructions using TaqMan Gene Expression master mix (Thermo Fisher Scientific) for 40 cycles. A target was defined to be present when a typical amplification curve was produced at a cycle threshold (ct) of fewer than 32 cycles. Predesigned Taqman probes were purchased from Thermo Fisher Scientific (Chrna1; Mm00431629_m1, Chrna2; Mm00460630_m1, Chrna3; Mm00520145_m1, Chrna4; Mm00516561_m1, Chrna5; Mm00616329_m1, Chrna6; Mm00517529_m1, Chrna7; Mm01312230_m1, Chrna9; Mm01221611_m1, Chrna10; Mm01274155_m1, Chrnb1; Mm00680412_m1, Chrnb2; Mm00515323_m1, Chrnb3; Mm00532602_m1, Chrnb4; Mm00804952_m1, Chrm1; Mm01231010_m1, Chrm2; Mm01167087_m1, Chrm3; Mm01338410_m1, Chrm4; Mm00432514_s1, Chrm5; Mm01701883_s1, Tubb3; Mm00727586_s1, Gfap; Mm01253033_m1). Non-template controls and a no-RT control were included. Cells with no Tubb3 (four cells) or positive Gfap (one cell) expression were excluded from analysis. A total of 48 cells were picked and analysed with 5 cells excluded.
Calcium imaging of small-intestine mucosa-innervating vagal ganglia neurons
Retrogradely traced vagal ganglion neurons were isolated from adult mice as previously described5. In brief, four days after retrograde tracing, mice were euthanized by CO2 inhalation, and vagal ganglia were surgically removed and digested as described above. Neurons were spot-plated onto coverslips coated with poly-d-lysine (800 mg ml−1) and laminin (20 mg ml−1) and maintained at 37 °C in 5% CO2. After 24 h in culture, neurons were loaded with 2.5 μM Fura-2-AM (Thermo Fisher Scientific) and 0.02% (v/v) pluronic acid for 30 min at room temperature in Ringer’s solution (NaCl 140 mM, KCl 5 mM, CaCl2 1.25 mM, MgCl2 1 mM, glucose 10 mM and HEPES 10 mM, pH 7.4). After a brief wash, coverslips were transferred to a recording chamber filled with Ringer’s solution at room temperature (around 22 °C). Retrogradely traced vagal ganglion neurons were identified by the presence of the AF488 tracer and viability was verified by responses to 40 mM KCl. Fura-2-AM fluorescence was measured at 340-nm and 380-nm excitation, and 530-nm emission was measured using an Olympus IX71 microscope in conjunction with a Sutter Lambda 10-3 wavelength switcher and the Chroma filter set no. 49011 (ET480/40x (Ex), T510lpxrxt (BS), ET535/50m (Em)). Fluorescence images were obtained every 5 s, using a 4× objective with a monochrome CCD camera (Retiga ELECTRO). Images were taken at baseline and after administration of m-chlorophenylbiguanide hydrochloride (mCPBG, 10 μM, Tocris), acetylcholine chloride (Sigma Merck) (10 μM) and KCl (40 mM). Fluorescence signals from neuronal cell bodies were extracted using MetaFluor software (Molecular Devices). ROIs were manually drawn around neuronal cell bodies and their fluorescence traces were extracted as the 340/380 ratio.
Analysis of single-cell and single-nucleus RNA-sequencing datasets
For mouse gut epithelial cells, we downloaded processed single-cell RNA-sequencing data (GSE224223)21 from the Gene Expression Omnibus (GEO) database, for which both tuft and enteroendocrine cell subtypes are well annotated. For the mouse nTS cell atlas, we downloaded processed single-nucleus RNA-sequencing data from wild-type mice (GSE200003)34 from the GEO, where each population is well annotated. All expression profiling was performed using modified DimPlot or VlnPlot functions from Seurat 5.0.1. (Satjia Laboratory, https://satijalab.org/seurat/) and ggplot2 on R v.4.4.1.
Statistical analysis
Data were analysed with Prism (GraphPad). n represents the number of mice, cells, crypts, villi, nerve fibres or independent experiments, as specified in the legends. Data were considered significant if P < 0.05. Statistical parameters are described in the figure legends. All significance tests were justified considering the experimental design, and we assumed normal distribution and variance, as is common for similar experiments. Sample sizes were chosen on the basis of the number of independent experiments required for statistical significance and technical feasibility.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
First Appeared on
Source link

