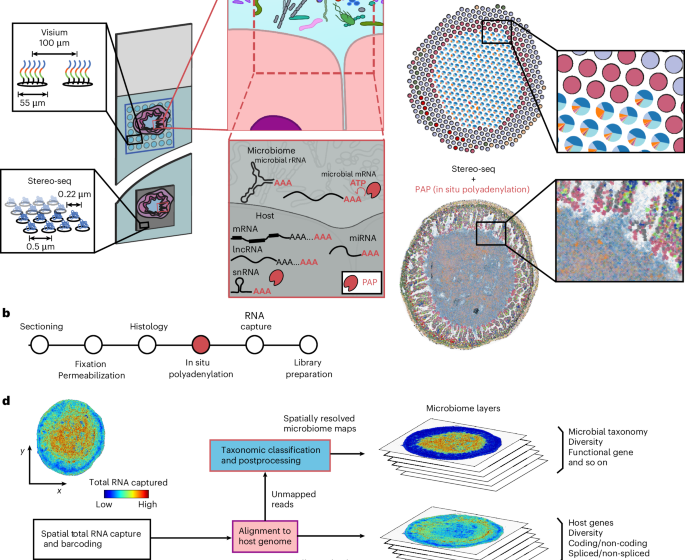
Spatial transcriptomics maps host–gut microbiome biogeography at high resolution
Animal models and experimental procedures
All animal protocols were approved by the Cornell University Institutional Animal Care and Use Committee (IACUC), and experiments were performed in compliance with institutional guidelines (protocol number IACUC 2016-0088). C57BL/6-ApcMin/+/J and C57BL/6 wild-type mice were used for the spatial transcriptomics experiments. All mice were maintained at the mouse facility at Weill Hall of Cornell University under standard housing conditions, including a 12 h light–12 h dark cycle, controlled ambient temperature (approximately 20–25 °C) and controlled humidity (approximately 30–70%), with ad libitum access to food (autoclavable Teklad global 14% protein rodent maintenance diet #2014-S; Envigo) and water. ApcMin/+ and wild-type mice were initially ordered from Jackson Laboratory and then bred in the barrier facility. The ApcMin/+ mice used in these experiments have a chemically induced transversion point mutation at nucleotide 2549, resulting in a stop codon at codon 850, truncating the APC protein.
Five mice were used for the spatial transcriptomics experiments: three C57BL/6-ApcMin/+/J (two female, one male) and two C57BL/6 wild-type mice (one female, one male), all approximately 13 weeks old at the time of collection. Overall health, food intake and weight of the mice were monitored to ensure tumour burden did not violate ethical standards. After approximately 100 days, mice were killed using 5 min of CO2 asphyxiation followed by tissue collection. Intestines from the mice were inspected for tumour localization, excess fat was removed, and cut into individual sections. Tissues were embedded in cryomolds with OCT compound (Tissue-Tek) and frozen in an isopentane-liquid nitrogen as previously described. The small intestine was cut into four to six approximately equal-sized segments and the large intestine into two to three segments, and the caecum was processed separately.
In situ polyadenylation reaction optimization
To determine an appropriate incubation time for in situ polyadenylation, an in vitro time-course assay was performed. A uniform 1.8 kb RNA library (Promega, L4561) was incubated with yeast poly(A) polymerase (yPAP, Thermo Scientific) under identical reaction conditions as the tissue sections. Duplicate reactions were prepared for a series of incubation timepoints (0, 1, 5, 15 and 25 min). Following polyadenylation, RNA was purified using a silica-membrane-based cleanup kit (RNA Clean and Concentrate, Zymo Research), and fragment size distributions were analysed with an Agilent Tapestation 4200. In bulk reactions, over 90% of input RNAs were polyadenylated within 1 min of incubation, and poly(A) tails exceeding 10 kb in length were detected after 15 min at 37 °C. On the basis of these results, a 25-min incubation was selected as adequate time for the enzymatic reaction in situ (Extended Data Fig. 10).
In situ polyadenylation for the gastrointestinal tract profiling with the Visium platform
Cryosections were obtained from the proximal small intestine, ileum, caecum and colon of a single individual (male, 13 weeks). Sections were processed using a polyadenylation protocol or the standard Visium protocol. For the polyadenylation protocol, 10-µm-thick tissue sections were mounted onto Visium Spatial Gene Expression v1 slides. Sections were fixed in freshly prepared methacarn solution (60% methanol, 30% glacial acetic acid and 10% chloroform) at room temperature for 15 min. H&E staining was performed according to the Visium protocol, and tissue sections were imaged using a Zeiss Axio Observer Z1 microscope equipped with a Zeiss Axiocam 305 colour camera. The resulting H&E images were corrected for shading, stitched, rotated, thresholded and exported as TIFF files using Zen 3.1 software (Blue edition). After imaging, slides were transferred into the Visium Slide Cassette. The methacarn fixation step serves as an initial permeabilization for the following enzymatic incubation.
In situ polyadenylation was conducted using yPAP (Thermo Scientific, catalogue number 74225Z25KU). Each capture area was equilibrated by adding 100 µl of 1× yPAP Reaction buffer (20 µl 5× yPAP reaction buffer, 2 µl 40 U µl−1 Protector RNase Inhibitor, 78 µl nuclease-free H2O), for 30 s at room temperature, after which the buffer was removed. Subsequently, 75 µl of yPAP enzyme mix (15 µl 5× yPAP reaction buffer, 3 µl 600 U µl−1 yPAP enzyme, 1.5 µl 25 mM ATP, 5 µl murine RNase inhibitor, 50.5 µl nuclease-free H2O) was added to each reaction chamber. The chambers were sealed and incubated at 37 °C for 25 min, and the enzyme mix was removed. Following polyadenylation, enzymatic permeabilization was performed for 30 min, followed by the standard Visium library preparation protocol to generate cDNA and final sequencing libraries. For the standard Visium experiment, polyadenylation was skipped and immediately proceeded to permeabilization.
In situ polyadenylation with the Stereo-seq platform
Adjacent ileal cross-sections to those profiled with Visium were profiled by STOmics platform. The 10-µm-thick sections were placed onto STOmics mini chips (product number 211ST004). For the polyadenylation protocol, sections were fixed in methacarn for 15 min, followed by a DNA staining step according to the STOmics protocol. Imaging was performed on a Zeiss Axio Observer Z1 Microscope using a Hamamatsu ORCA Fusion Gen III Scientific CMOS camera. Images were stitched, rotated, thresholded, processed and exported as TIFF files using Zen v.3.1 software (Blue edition), then registered using the STOmics software. After imaging, in situ polyadenylation was performed followed by 12 min permeabilization and library preparation. For the standard experiment, polyadenylation was skipped and immediately proceeded to permeabilization.
Additional samples profiled included ileal sections from two mice, male and female, with a tumour adjacent to the lumen, four sections from a healthy C57BL/6 wild-type male (proximal small intestine, ileum, caecum and distal colon) and one proximal colon section from a wild-type female.
Sequencing of the spatial transcriptomics libraries
Sequencing of the Visium libraries was performed on a NextSeq 2K (Illumina) platform using a P3 200 bp kit, with reads allocated as follows: 28 bp for read 1, 10 bp for index 1, 10 bp for index 2 and 190 bp for read 2. For the libraries prepared using the STOmics platform, sequencing was carried out on a Complete Genomics DNBSEQ-T7 Sequencer using the DNBSEQ-T7 High-throughput Sequencing Set (FCL PE100) and the associated STOmics primer set. The sequencing run consisted of a 50-bp read 1 (with dark cycles from bases 26 to 40), a 100-bp read 2 and a 10-bp index read.
Metatranscriptome library preparation of adjacent sections and sequencing
For each intestinal region, adjacent cryosections corresponding to Visium and Visium + PAP samples were processed for bulk metatranscriptomic analysis. Total RNA was extracted using the NucleoSpin RNA Stool Kit (Takara Bio, 740130.50). To remove contaminating genomic DNA, DNase treatment was performed using a two-step protocol: (1) initial treatment with Baseline-ZERO DNase (1 μl; LGC Biosciences, DB0715K) and (2) treatment with TURBO DNase (3 μl; Thermo Fisher Scientific, AM2238) in 10 μl of the supplied buffer. A 1-μl aliquot of RNA was diluted in 99 μl of nuclease-free water, incubated at 37 °C for 30 min, and then placed on ice. RNA cleanup was performed using the RNA Clean and Concentrator kit (Zymo Research, R1013), and eluted in 10 μl of nuclease-free water.
Stranded total RNA-seq libraries were prepared using the SMARTer Stranded Total RNA-Seq Kit v3—Pico Input Mammalian (Takara Bio, 634485), following the manufacturer’s protocol. The four libraries were pooled and sequenced with MiniSeq sequencer (Miniseq High Output Reagent Cartridge, 150 Cycles, Ilumina).
Preprocessing and alignment of spatial transcriptomics data
To ensure similar alignment and quantification across platforms and methodologies we used the ‘slide_snake’ pipeline that utilizes Snakemake53 (6.1.0), which can be found on GitHub54. For the Visium and STRS (Visium) libraries, the pipeline first trims poly(A) and poly(G) sequences, and primer sequences using cutadapt55. The reads were aligned using STAR v2.7.10a56 and STARSolo57 (specified parameters: –outFilterMultimapNmax 50, –soloMultiMappers EM, –clipAdapterType CellRanger4) to generate expression matrices for every sample. The GeneFull matrices were used for downstream analyses. Barcode whitelists and the associated spot spatial locations for Visium data were copied from the Space Ranger software (‘Visium-v1_coordinates.txt’). For the Stereo-seq and STRS (Stereo-seq) libraries, barcode maps were provided by the manufacturer as.h5 files and converted to text format using ST_BarcodeMap58. Alignment references were generated from the GRCm39 reference sequence using GENCODE M32 annotations.
Unmapped reads classification and construction of microbiome Anndata objects
In this study, Kraken2 (version 2.09)22 was used to classify microbial reads. We used the standard Kraken2 database supplemented with the mouse genome. Unmapped reads flagged in the BAM file were processed to retain the correct cell barcode and UMI information as identified by STARsolo. This permitted the demultiplexing of Kraken2 output by cell barcode and UMI. For data integration, we employed Pandas, Scanpy, NumPy, Scipy and regular expressions to create an AnnData object with cell barcodes as observations and NCBI taxonomy IDs as features. Only classified reads were retained for subsequent analysis.
Image registration and cell segmentation
For Visium and Visium + PAP samples, image registration was performed using the 10x Genomics Loupe Browser. H&E-stained tissue images were aligned to the spatial capture array, and tissue and lumen regions were manually annotated. For Stereo-seq samples, nuclear-stained fluorescence images were acquired during sample processing. Image registration and cell segmentation were carried out using the Stereo-seq Analysis Workflow (SAW) provided by STOmics. Fluorescence images were aligned to the chip layout based on the ChipID metadata. Following registration, automated cell segmentation was performed using SAW’s built-in algorithms. Segmentation masks were used to define cell boundaries, and barcodes within each segmented region were aggregated to construct single-cell transcriptomes. These cell-level datasets were then used for downstream spatial analysis and deconvolution.
Processing and alignment of metatranscriptomic libraries
Metatranscriptomic sequencing data were processed using a custom computational workflow. Adaptor trimming and quality filtering were performed using BBDuk (v38.90). Filtered reads were aligned to the mouse genome (GRCm39) with STAR (v2.7.10a) using GENCODE M32 annotations. Gene-level quantification was carried out with featureCounts (v2.0.0). Duplicate reads were identified and marked using Picard MarkDuplicates (v2.19.2). Unmapped reads after host genome alignment were extracted and taxonomically classified using Kraken2 (v2.0.9).
Sterile control preprocessing and identification of taxa to filter
To assess the Kraken2-classified microbial counts occurring in non-intestinal tissues for the low-resolution platform we realigned previously published Visium and STRS libraries of mock-infected C57BL/6J 11-day-old mice with and without polyadenylation as described in the corresponding studies15,21. A total of 85 taxa occurring at 1 ppm (UMI) or greater were excluded from downstream analysis as potential misclassification. For the Stereo-seq libraries, a sterile control experiment was conducted. In brief, fresh-frozen heart from an 11-day-old mouse was sectioned on a Stereo-seq 1 cm × 1 cm tile (STOmics, BGI). The sample was fixed in methanol at −20 °C for 20 min followed by the in situ polyadenylation and the Stereo-seq library preparation protocol as described above. Taxa occurring at frequencies higher than 1 ppm UMI were excluded from downstream analyses.
Preprocessing of the Visium and Visium + PAP data
Spatial coordinates were assigned to the Visium and Visium + PAP library spots based on the barcode map provided by the Space Ranger software (‘Visium-v1_coordinates.txt’). The accompanying H&E histology images of each experiment were used to manually mark the spots that correspond to tissue and lumen. Scanpy59, mudata60,61 and muon60 were used to construct multimodal objects separately for the microbial maps (in the taxonomic levels of phylum, family, genus and species). This was done for each one of the accounted microbial superkingdoms of Archaea, Bacteria and Viruses. For downstream analyses, only the spots covered by tissue or corresponding to lumen were accounted for.
Microbial percentage and enrichment calculation for the paired Visium and Visium + PAP experiments
Microbial percentages were calculated as the fraction of Kraken2-classified unique molecules assigned to each superkingdom, normalized by the total library size (host-aligned plus Kraken2-classified unique molecules). Enrichment was defined as the ratio of those percentages between paired Visium + PAP and standard Visium experiments.
Relative abundance and bacterial richness calculations for the low-resolution datasets
To calculate the relative abundance for each sample, at the family level, the corresponding family reads were collapsed and divided by the total molecules originating from bacteria as classified by Kraken2. The microbial richness per spot was calculated as the number of unique taxa occurring per spot after the exclusion of taxa accounting for 0.01% or less of microbial molecules in the whole sample. For the transverse axis relative abundance analysis, spots were spatially binned from the tissue to the lumen based on their minimum distance to the lumen-associated region. Phyla relative abundance data were then aggregated within each bin to quantify relative abundances across the tissue–lumen axis.
Rarefaction and sequencing saturation analysis
Rarefaction analysis was performed on paired Visium and Visium + PAP libraries collected from the four intestinal regions. Sequencing data were subsampled at defined fractions (10% to 100% of total reads) and aligned to the mouse genome using the Snakemake-based pipeline. The unmapped reads were extracted and taxonomically classified using Kraken2 and AnnData objects were generated as described for the full dataset. For classified microbial reads in each condition, rarefaction curves were generated by plotting the number of unique bacterial molecules against sequencing depth. Michaelis–Menten models were fitted using nonlinear least squares to estimate theoretical saturation behaviour, and model fit was evaluated using the coefficient of determination (R²). To calculate saturation for the total library, and for host- and microbiome-derived reads, the following general formula was used:
$$\mathrm{Saturation}=1-\frac{\mathrm{Modality}\,\mathrm{unique}\,\mathrm{molecules}}{\mathrm{Modality}\,\mathrm{reads}\,\mathrm{with}\,\mathrm{valid}\,\mathrm{barcode}}.$$
Here, modality refers to the source of molecules: host-aligned reads, microbial reads classified by Kraken2 or all reads combined. This metric captures diminishing returns in the recovery of unique molecules with increasing sequencing depth. Saturation was computed at each subsampling level and modelled using Michaelis–Menten kinetics constrained to a maximum of 1. The resulting curves were used to estimate the sequencing depth required to achieve saturation (for example, saturation of ~0.9).
Gram stain comparison of Visium + PAP to the bulk RNA measurement
Adjacent samples profiled by Visium + PAP and by bulk metatranscriptomics were compared at the genus level. Reads were taxonomically classified with Kraken2 and counts were aggregated by genus. The same taxa excluded in spatial control analyses were removed before comparison. Gram-stain labels for genera were retrieved from BacDive62 and were curated to fill missing entries for highly abundant genera, including the Gram-negative: Pseudoprevotella, Hoylesella, Segatella, Allomuricauda, Marvinbryantia, Lachnoclostridium, Vescimonas, Mediterraneibacter, Coprococcus, Caproicibacterium, Massilistercora, Tellurirhabdus, Blattabacterium, Koleobacter and Faecalitalea. The labels were used to calculate percent Gram-positive, Gram-negative, unknown or unclassified for the two types of measurement.
Cell type deconvolution
We employed the cell2location28 model (version 0.1.3) to deconvolve spatial transcriptomics generated with the Visium and Stereo-seq platforms. The scRNA-seq reference, from a previous study on Apc Min/+ mice27 was filtered to include only genes that are highly expressed and informative for identifying rare cell types, with thresholds set at cell_count_cutoff of 5, cell_percent_cutoff of 0.01 and nonz_mean_cutoff of 1.12. Cell-type-specific expression signatures were generated using negative binomial regression from these selected genes and applied to the spatial transcriptomics data to determine cell-type identities, with the highest prediction scores used for assignment. The detection_alpha parameter was set to 20 and N_cells_per_location was set to 30 for Visium and 1 for Stereo-seq.
Bacterial gene function analysis
Bacterial reference resources comprised selected abundant bacterial genomes (accession IDs GCF_037113525.1 and GCF_025148285.1) and bacterial genes downloaded from NCBI (downloaded 18 September 2025); for the genes, we retrieved and used all sequences annotated under the names atpA, enolase, tufA, eno, gap, groL, msmX, pckA, ppdK and spoIVCA. Whole-genome FASTA files were annotated with Prokka (v[1.14.5]) using default parameters to produce GFF3 feature annotations (genes, rRNA/tRNA, CDS and product fields). Unmapped reads from the alignment to the host genome were mapped to the bacterial references using Bowtie2 (v[2.5.1]) with default parameters after building indexes via bowtie2-build. Genome-wide mapping summaries were visualized with pycirclize (v[1.6.0]) as Circos-style plots. Tracks included (1) annotated ribosomal RNA/protein genes from the Prokka GFF, (2) optional GC/AT content and (3) 1-kb binned coverage. Values used for plotting were clipped to the predefined display range to avoid axis boundary artefacts. The per-base depth was computed from sorted BAMs with samtools depth -aa, then aggregated into non-overlapping 1-kb windows (mean per window) and written as bedGraph (chrom, start, end, mean). Using these bins (midpoints as genomic positions), we optionally smoothed coverage (window of 1 bin; none) and called peaks with scipy.signal.find_peaks (prominence of 0.2 × 10th percentile of positive bins; minimum distance of 5 bins is ~5 kb). Prokka GFF3 annotations (gene/CDS/rRNA/tRNA) were parsed, and multisegment features with the same name were merged (minimum start, maximum end). Each peak was labelled overlap if its bin intersected any feature (all overlaps recorded, one representative chosen) or nearest otherwise (nearest feature and distance reported)63,64.
Spatial autocorrelation analysis
Moran’s I was calculated for the major genera (abundance >0.01%) using the Moran function from the Python library pysal. Spatial weights were generated using the k-nearest neighbours matrix (k = 4) from the weights module in pysal. For genera with a Moran’s I P value <0.05, Ripley’s H was subsequently derived using the following formula:
$$K\left(r\right)=\frac{2A}{N\left(N-1\right)}\mathop{\sum }\limits_{i=1}^{N}\mathop{\sum }\limits_{j=i+1}^{N}I\left({d}_{{ij}}\le r\right)$$
$$L(r)=\sqrt{\frac{K(r)}{{\rm{\pi }}}}$$
where \({d}_{{ij}}\) is the Euclidean distance between points i and j. \(I({d}_{{ij}}\le r)\) is an indicator function that is 1 if the distance \({d}_{{ij}}\) is less than or equal to \(r\) and 0 otherwise. \(A\) is the area of the observation window. \(N\) is the number of points in the dataset.
Spatial colocalization analysis
To assess the spatial colocalization of bacterial genera in the Stereo-seq datasets, we applied the Smoothie package using default parameters for the Stereo-seq platform30. In brief, bacterial spatial data classified at the genus level by Kraken2 were spatially smoothed (20-μm Gaussian kernel), and pairwise correlations were computed among all resulting bacterial signal surfaces to quantify colocalization patterns across the tissue area.
Spatial clustering of microbial signal (HDBSCAN65) and contouring
To assess the spatial clustering characteristics of a given bacterial taxa, we subset spots with nonzero counts and used their spatial coordinates (obsm[‘spatial’]) as input to HDBSCAN (v[0.8.38]). To choose hyperparameters, we performed a grid search over min_cluster_size and min_samples. For each setting we fit HDBSCAN, removed noise labels (−1) and clusters with size
Boundary detection
The microscope data were saved in greyscale and then averaged using the OpenCV blur function with a kernel size 100 µm. After that, the data were binarized with a threshold of 80 for normal tissue and 100 for cancer tissue. Finally, boundaries were extracted using the OpenCV findContours function.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
First Appeared on
Source link

