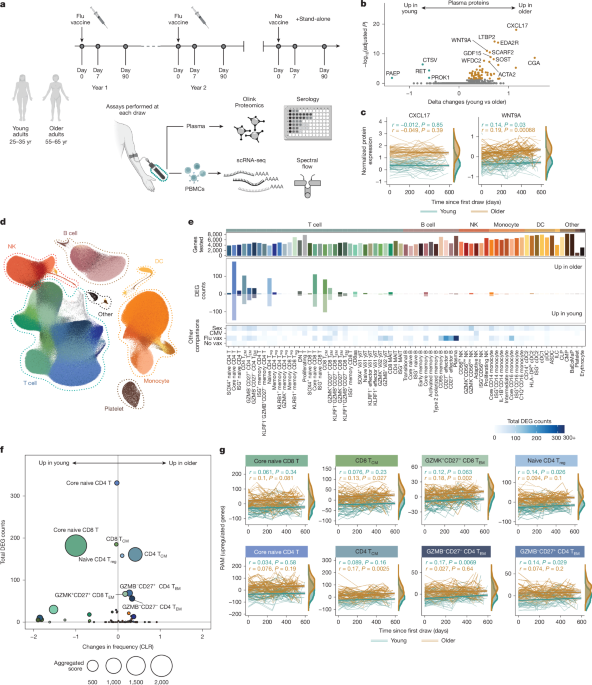
Multi-omic profiling reveals age-related immune dynamics in healthy adults
The Sound Life study cohort
Healthy 25- to 35-year-old and 55- to 65-year-old adult donors were prospectively recruited from the greater Seattle, Washington, USA, area as part of the Sound Life Project, a protocol (IRB19-045) approved by the Institutional Review Board (IRB) of the Benaroya Research Institute. All adult participants provided informed consent before participation. Donors were excluded from enrolment if they had a history of chronic disease, autoimmune disease, severe allergy or chronic infection. All blood samples were collected, processed to PBMCs through a Ficoll-based approach and frozen within 4 h of blood draw. Plasma samples were processed, aliquoted and frozen within 4 h of blood draw. Basic demographics are provided in Supplementary Table 1. Extensive clinical data were collected on donors at each blood draw and is available for detailed exploration at https://apps.allenimmunology.org/aifi/insights/dynamics-imm-health-age/vis/clinical/. Donors received the following inactivated, quadrivalent influenza vaccinations based on the corresponding flu season; FLUARIX Quadrivalent in 2019-2020 and 2021–2022 and Fluzone in 2020–2021.
Follow-up cohort
Under a protocol approved by the Stanford University IRB, 234 paired PBMCs and serum samples were retrospectively selected from a cohort of healthy adults, ages 40 years and older, recruited from the greater Palo Alto, California, USA area2. All participants provided informed consent before participation and these subsequent studies were approved by the Allen Institute IRB. Basic demographics for the selected follow-up cohort are provided in Supplementary Table 1.
Olink Explore
The Olink Explore 1536 platform was used for the Sound Life cohort plasma samples to measure relative expression of 1,472 protein analytes. The Olink Explore 3072 platform was used to measure expression of 2,943 protein samples in serum samples of the follow-up cohort. Samples were randomized across plates to achieve a balanced distribution of age and sex. Bridging controls were used in each run for cross-batch normalization.
scRNA-seq via 10x Genomics
scRNA-seq was performed on thawed PBMCs as previously described50. Chromium 3′ v.3 chemistry was used for scRNA-seq of the Sound Life cohort. In brief, thawed PBMCs were stained for oligonucleotide-tagged antibodies (HTO) and overloaded on Chip G wells (10x Genomics, 20000177) at 64,000 cells. cDNA amplification was done with HTO additive primer spiked in the master mix. HTO and GEX products were then separated using SPRI-Select (Beckman Coulter, B23319) bead-based cleanup before carrying forward into separate library indexing reactions. Chromium Fixed RNA profiling was used for scRNA-seq of the follow-up cohort with bridging controls included for cross-batch normalization downstream. In brief, up to two million cells per sample were processed (10x Genomics, 1000414) and probes were hybridized (10x Genomics, 1000476) per the manufacturer’s instructions. Samples were pooled and loaded onto two wells of Chip Q (10x Genomics, 2000518) for gel beads-in-emulsion generation, and subsequent library generation. Libraries were sequenced using a NovaSeq X 25B 300 cycle flow cell or NovaSeq S4 200 cycle flow cell, depending on total read requirements at either Clinical Research Sequencing Platform at the Broad Institute (https://broadclinicallabs.org/) or Northwest Genomic Center at the University of Washington (https://nwgc.gs.washington.edu/). Samples were then computationally resolved and quality-checked using in-house pipelines.
Human CMV serology
Viral serology testing for human CMV was performed at the University of Washington’s Clinical Virology Laboratory in the Department of Laboratory Medicine (https://depts.washington.edu/uwviro/). Plasma or serum samples (200 µl) were run through the FDA-approved LIAISON CMV IgG Assay to qualitatively detect CMV IgG class antibodies. Results were reported for each sample as ‘positive’ or ‘negative’ along with a CMV antibody screen index value ranging from <0.20 to >10.00.
Adaptive NK cell flow cytometry
Between 1 and 2 million PBMCs seeded in 96-well U-bottom plates were stained for viability, Fc blocked, and then stained with a surface marker antibody cocktail (Supplementary Table 4) using BD Brilliant Staining Buffer (BD Biosciences, 563794) for 30 min at 4 °C. The samples were then washed, fixed for 60 min at room temperature, Fc blocked and permeabilized for 10 min at room temperature. Cells were then incubated with an intranuclear antibody staining cocktail using the eBioscience Foxp3/Transcription Factor Staining Buffer Set (Thermofisher, 00-5523-00) for 60 min at 4 °C, washed, fixed, washed again, resuspended and acquired on a BD Symphony (5L) flow cytometer. Data were collected by using BD FACSChorus (v.2).
HAI influenza-specific antibody serology
The Meso Scale Discovery (MSD) 96-well HAI 9-plex Assay measures neutralizing antibodies in human plasma that block the binding of labelled red blood cell vesicles to trimeric Influenza HA antigens, specific for the following lineages: A/Brisbane, A/Cambodia, A/Guangdong, A/HongKong, A/Kansas, A/Shanghai, A/Wisconsin, B/Phuket and B/Washington. In brief, MSD 96-Well 10-Spot multi-array plates coated with 9 trimeric flu HA antigens were blocked and then pretreated human plasma samples (to remove sialic acid residues) diluted 5,000-fold along with HA reference standards were added to the plate. Plates were shaken for 2 h at 15 °C to 25 °C, ECL SULFO-TAG labelled red blood cell vesicles were added, shaken again for 2 h, washed, MSD GOLD Read Buffer B was added, and then plates were read on an MSD SECTOR S600 ECL plate reader. Test samples were reported as per cent inhibition, relative to a no-plasma diluent only control. Positive samples show high per cent inhibition whereas negative or low samples show low per cent inhibition.
B cell flow cytometry
PBMCs were rapidly thawed, seeded at 2 million cells per well in a 96-well plate, and incubated with Human TruStain FcX and Purified mouse IgG (BioRad, PMP01X) for 10 min at room temperature. Cells were incubated with Fixable Viability Stain 510 (BD Biosciences, 564406) for 30 min at 4 °C, washed, and stained with an antibody cocktail for 30 min at room temperature (Supplementary Table 4). After another wash, the cells were resuspended and acquired on an Aurora 5 L flow cytometer (Cytek Biosciences). Data are collected by using Cytek SpectroFlo software (v.2.0.2).
Total influenza-specific antibody serology
The MSD Prototype Influenza 7-plex Serology Assay protocol measures IgG antibodies in human plasma specific for Influenza vaccine HA antigens: A/Brisbane, A/Hong Kong, A/Michigan, A/Victoria, B/Colorado, B/Phuket and B/Washington. In brief, MSD 96-Well 10-Spot multi-array plates coated with seven flu HA antigens were blocked, human plasma samples were diluted 10,000-fold, and added along with HA reference standards and controls to the plate. Plates were shaken for 2 h at 15 °C to 25 °C, washed, anti-human IgG antibodies labelled with electrochemiluminescent (ECL) SULFO-TAG added, shaken again for 1 h, washed, MSD GOLD Read Buffer B added, and read on an MSD SECTOR S600 ECL plate reader. Test samples were quantified in absorbance units per ml (AU ml−1), referenced against specific HA reference standards.
MSD influenza 7-plex IgG subtype serology assay
This assay protocol is for measuring IgG1, IgG2, IgG3 and IgG4 subtype antibodies in human plasma specific for Influenza vaccine HA antigens: A/Brisbane, A/Hong Kong, A/Michigan, A/Victoria, B/Colorado, B/Phuket and B/Washington. In brief, MSD 96-Well 10-Spot multi-array plates coated with 7 flu HA antigens were blocked, then human plasma samples diluted 10,000-fold, HA reference standards, and controls were added to the plate. Plates were shaken for 2 h at 15 °C to 25 °C, washed, then unconjugated mouse anti-human IgG1, IgG2, IgG3, or IgG4 monoclonal antibodies (Southern Biotech) added, shaken again for 1 h, washed, goat anti-mouse IgG labelled with ECL SULFO-TAG added, shaken again for 1 h, washed, MSD GOLD Read Buffer B added, and read on an MSD SECTOR S600 ECL plate reader. Test samples were quantified in AU ml−1, referenced against specific HA reference standards.
Cytokine production by T cells
Cell supernatants from stimulated and unstimulated cells were analysed for cytokine production using the MSD platform. Seven cytokines (IL-1B, IL-4, IL-6, IL-8, IL-10, TNF and IFNy) were measured using a chemiluminescence-based assay from MSD (N05049A-1) according to manufacturer’s instructions. Serial fourfold dilutions of supernatants, 1:8 and 1:32 for 4 h and 24 h samples, respectively, were prepared in Diluent 2 for conformity with the limits of detection. Analyses were done using the Discovery Workbench 4.0 software and R studio v.4.4.2.
TH cytokine and CD40 ligand flow cytometry
Cryopreserved PBMCs were rapidly thawed, and T cells were enriched using EasySep Human T Cell Isolation Kit (17951; Stemcell Technologies) and plated at 500,000 cells in medium alone or 250,000 cells with Dynabead Human T-Activator CD3/CD28 Beads (11131D; Thermo Fisher) for 4 h or 24 h. Golgi plug was added 4 h prior to collection. Cells were then stained with a surface marker panel, fixed, washed, permeabilized and then stained with intracellular antibodies. Samples were collected on a Cytek 5L Aurora and analysis was performed using FlowJo v.10.10 software.
AIFI Immune Health Atlas
To build our scRNA-seq PBMCs dataset for the AIFI Immune Health Atlas, we utilized data and analysis environments within the Human Immune System Explorer (HISE) system (https://allenimmunology.org/) to trace data processing, analytical code and analysis environments from the original, raw FASTQ data to our final, assembled reference atlas51. A graph representation of the analysis trace for this project is available at https://apps.allenimmunology.org/aifi/resources/imm-health-atlas/reproducibility/. Additional details are available in our analysis notebooks on Github at https://github.com/aifimmunology/aifi-healthy-pbmc-reference and at our website at https://apps.allenimmunology.org/aifi/resources/imm-health-atlas/.
Data selection from HISE storage
To assemble the input data for our reference dataset, we selected samples from all donors in our longitudinal cohort (Sound Life cohort; young adult, age 25–35 years and older adult, age 55–65 years) and 16 samples from a previously described healthy paediatric cohort collected at the University of Pennsylvania (age 11–13 years)32. In total, 108 samples were selected for use in our reference (paediatric, n = 16; young adult, n = 47; older adult, n = 45), consisting of 2,093,078 cells before additional quality control (QC) filtering.
Pipeline processing
After sequencing, gene expression and Hash Tag oligonucleotide libraries from pooled samples in our pipeline batches were demultiplexed and assembled as individual files for each biological sample as previously described52.
Cell labelling
To guide cell-type identification, we labelled cells from each sample using CellTypist (v.1.6.1)53, using the following reference models: Immune_All_High (32 cell types), Immune_All_Low (98 cell types), and Healthy_COVID19_PBMC (51 cell types) by following the approach described in the CellTypist reference documentation at https://celltypist.org. We also labelled our cells using Seurat (v.5.0.1)54 (https://zenodo.org/doi/10.5281/zenodo.7779016), first transforming to reference dataset using SCTransform followed by FindTransferAnchors and MapQuery to assign cell types.
Doublet detection and QC filtering
After assembly of all cells across 108 samples, we removed possible doublets (based on scrublet and high gene detection, >5,000 genes), low-quality cells (based on low gene detection, <200 genes), and dying or dead cells (based on mitochondrial unique molecular identifier (UMI) content, >10%). Doublet detection was done using the scrublet package (v.0.2.3)55. Mitochondrial content was assessed using the scanpy function calculate_qc_metrics (https://scanpy-tutorials.readthedocs.io/en/latest/pbmc3k.html). In total, 140,950 cells were removed (6.73%), and 1,952,128 cells passed QC filtering (93.27%).
Clustering and cell subsetting
After QC filtering, all remaining cells were clustered using a Scanpy workflow56 to normalize, log transform, perform principal components analysis (PCA), integrate age groups with Harmony57, perform Leiden clustering58, and generate two-dimensional UMAP projections. Clusters were assigned to major cell classes on the basis of marker gene detection, using the scanpy function scanpy.tl.rank_genes_groups. For each cluster, the fraction of cells with detected gene expression for each marker was computed, and clusters meeting a set of cell class-specific criteria were selected for downstream annotation by our domain experts. Clusters from specific cell classes were selected from the full dataset for iterative clustering analysis where necessary to identify cell subpopulations with additional resolution.
Expert annotation of cell types
Following high-level and iterative clustering, teams of domain experts within the Allen Institute for Immunology examined marker gene expression for clustered datasets and assigned cell-type identities to each cluster, also removing low-quality/doublet clusters, nine low-resolution cell classes (AIFI_L1), 29 mid-level cell classes (AIFI_L2), and 71 high-resolution cell classes (AIFI_L3) were identified, and each cell was labelled accordingly.
Training cell-type labelling models
In order to utilize our cell-type atlas to label other PBMCs datasets, we used CellTypist (v.1.6.2)53 to generate cell-type labelling models. We used a slightly modified CellTypist v.1.6.2 (allowing multinomial method: LogisticRegression(), alongside One-vs-Rest (OvR)), available at, labels. We utilized OvR regression for AIFI_L1 and AIFI_L2 labels, and Multinomial regression for AIFI_L3.
Sound Life scRNA-seq dataset assembly
To build our longitudinally sampled PBMC scRNA-seq dataset, we utilized data and analysis environments within the HISE system to trace data processing, analytical code, and analysis environments from raw FASTQ data to a labelled, high-quality final dataset51. A graph representation of the analysis trace for this project is available at https://apps.allenimmunology.org/aifi/insights/dynamics-imm-health-age/reproducibility/. Additional details are available in our analysis notebooks on Github https://github.com/aifimmunology/sound-life-scrna-analysis/ and at our website https://apps.allenimmunology.org/aifi/insights/dynamics-imm-health-age/.
Data selection
Input data were assembled as described above for the PBMC Atlas dataset. In total, 868 samples were selected for use in our dataset (young adult, n = 49 donors; n = 418 samples; older adult, n = 47 donors, n = 450 samples), consisting of 15,781,886 cells before additional QC filtering.
Pipeline processing
After sequencing, pipeline processing was done as described above for the PBMC Atlas dataset.
Labelling and doublet detection
To add cell-type labels, we utilized CellTypist (v.1.6.1) and the CellTypist models we generated using our Immune Health Atlas dataset at three levels of cell-type resolution (AIFI_L1; AIFI_L2, and AIFI_L3) per the reference document (https://celltypist.org).
QC filtering
After assembly of all cells across our samples, we filtered our data against the same set of QC criteria as described above for the PBMC Atlas. In total, 951,864 cells were removed (6.03%) and 14,830,022 cells passed QC filtering (93.97%).
Clustering, subsetting and doublet removal
All remaining cells were clustered within AIFI_L2 labels using the scanpy workflow described above for the PBMC Atlas. Clusters were filtered to remove doublets if multiple different cell-type marker genes were expressed, or if the cluster had low gene detection (low quality). Of note, due to their generally lower gene detection, this threshold was not applied to Erythrocyte or Platelet populations. In total, 800,059 cells (5.39%) were removed. 14,029,963 cells (94.61%) were retained for final cleanup based on high-resolution AIFI_L3 labels.
Refinement of cell-type labels
We worked with the high-resolution AIFI_L3 labelling results. For each cell type, we performed the scanpy data processing procedure described above to enable a final review of cell-type labels, and removed any doublet clusters. 234,117 cells were removed, and 13,795,846 were retained for the final reference dataset and downstream analyses.
Follow-up cohort scRNA-seq reference
Using our Atlas dataset, all cells in the follow-up cohort were automatically annotated with custom label transfer models built using the CellTypist framework. First, the genes in the reference dataset were subsampled to match the 18,000 genes in the 10x FLEX scRNA-seq assay. Then, three models were built for label transferring—one for each cell labelling level. Finally, the follow-up cohort data were labelled. Dataset cleanup, quality checking, clustering, and visualization were carried out using Scanpy. Doublets were detected and removed using Scrublet, and minimal inter-batch effects were adjusted in the PCA space for visualization purposes using Harmony. After QC filtering, labelling and data cleanup, 3,627,005 cells were retained in the final follow-up reference dataset and used for downstream analyses.
Olink analysis
Olink data normalization was first done using an internal extension control (IgG antibodies conjugated with a matching oligonucleotide pair). Next, they were normalized to the inter-plate pooled serum or plasma controls and finally, they were intensity normalized across all cohort samples. They are reported as log2(NPX) values. Using lme4 package in R59, the design formula: NPX(bridged) ~ age group + CMV + sex, was applied, with comparisons on the age group factor. Proteins with an adjusted P value below 0.05 were considered differentially expressed.
Milo differential abundance testing
We used milopy with default settings to perform abundance testing. For the scRNA reference, we conducted age (child versus older) and CMV status (negative versus positive) comparisons. For the flu vaccination comparison (day 0 versus day 7), we only used population from memory B cells populations and plasma cells.
Cell frequency comparison
To address the constant sum issue in compositional data, we applied a CLR transformation to frequency data from 71 level 3 cell types for each scRNA-seq sample. Of note, for switched versus non-switched CD27− effector B cells, the CLR was based on total memory B cells (level 3). In flow cytometry comparisons, the CLR was based on total T cells (TFH data) or 9 B cell types plus 1 non-B cell type (B cell data). For B cell isotype analysis, CLR was performed per cell type across isotypes. Paired data were analysed with the signed-rank Wilcoxon test; unpaired data with the rank-sum Wilcoxon test. For CMV-specific permutation analysis, we performed 100,000 label permutations of CLR-transformed frequencies. Median differences by cell type and CMV group formed a null distribution. Two-sided P values were computed by comparing observed versus permuted absolute differences, with a + 1 correction to avoid zero P values.
DEG analysis
Differential gene expression analysis was performed using the DESeq2 (v.1.42.0)60 package in R (v.4.3.2). For all comparisons, genes were filtered on the basis of a minimum of 10% non-zero counts in each comparison, to reduce noise, inconsistently expressed genes and technical artefacts in the DEG analysis, while balancing potential trade-offs between sensitivity and specificity61,62,63. Aggregated counts from single cells were used for the comparisons. The following design formulas and contrasts were used for the analyses:
-
1.
Baseline comparing flu year 1 day 0 samples. Design: aggregated counts ~ age group + CMV + sex. Contrast: each factor.
-
2.
Flu vaccine versus no-vaccine of year 1 and year 2. Design: aggregated counts ~ visit + age group + CMV + sex + subject. Contrast: visit factor (day 0 versus day 7).
-
3.
Flu response comparison between two age groups (aligned day 7 samples from 2020–2021 and 2021–2022 flu seasons). Design: aggregated counts ~ flu year + age group + CMV + sex. Contrast: age group factor (young versus older).
-
4.
CMV comparison. Design: aggregated counts ~ age group + CMV + sex. Contrast: CMV factor (positive versus negative).
Genes were considered differentially expressed if their adjusted P value was below 0.05 and their absolute fold change was greater than 0.1.
IMM-AGE score calculation
To calculate the IMM-AGE score, we first extracted the frequencies of cell types that significantly differed (Padj < 0.05) between the young and older groups by comparing scRNA-seq data from year 1, day 0 samples. We then applied dimensionality reduction using DiffusionMap function from the destiny package on the scaled frequency matrix and selected the second diffusion component as our IMM-AGE score.
Flow cytometry data analysis and visualization
T cell, NK and B cell flow cytometry data analysis consisted of traditional hierarchical gating in FlowJo v.10.10 Software. Gating strategies are provided in Supplementary Figs. 1–4. For deeper B cell and CD3+ T cell analysis, flow data were transformed (inverse hyperbolic sine with a cofactor of 220) and scaled (99.9th percentile of expression of all cells) in R. Total live B cells were over-clustered into 100 clusters using FlowSOM with all informative surface molecules as input. Clusters were then hierarchically clustered on the basis of expression of B cell surface molecules and isotype and manually assigned to cell subsets, as described previously25. UMAPs of B cell subsets were plotted using all informative markers and excluding isotype.
Enrichment analysis
Two types of enrichment analysis were conducted. For Gene Set Enrichment Analysis (GSEA), genes were ranked by −log10(P value) × sign(log2(fold change)) from DESeq2 results, and enrichment was run using the fgsea R package (v.1.28.0). Pathways with adjusted P values < 0.05 were retained. For sample level enrichment analysis (SLEA), genes above a 10% expression threshold served as the background. Pathway scores were calculated using the leading-edge genes from the fgsea results. At the pseudobulk level, we calculated the mean gene expression with random genes from the background set for each sample, performing 1,000 iterations. The pathway score was defined as the z-score of this difference.
Response score and durability score definition
The HAI data were normalized by the minimum value per batch and assay, added 0.00001 to ensure positive and non-zero values, and fitted with a linear model to estimate response/durability scores for each strain by each flu year separately. Residuals from the models were taken as individual response scores. Response score model: Δ(day 7 – day 0) HAI inhibition ~ day 0 inhibition + sex + CMV. Durability score model: Δ(day 90 – day 7) HAI inhibition ~ day 7 inhibition + sex + CMV.
NMF projection for TH state score determination
We used the NMF projection tool (https://github.com/yyoshiaki/NMFprojection/tree/main) with its precomputed NMF matrix (https://github.com/yyoshiaki/NMFprojection/blob/main/data/NMF.W.CD4T.csv.gz) to compute the NMF scores on Python (v.3.10.13), using the recommended workflow for subsetted CD4 T cells in each sample. Obtained NMF scores were normalized across each cell type at the sample level. For the top genes defining each NMF factor, we extracted the weight of each gene from the precomputed NMF matrix. Genes with higher weights were considered to be more contributive to each factor. We choose the top 20 genes for TFH NMF factor with highest weight.
Cell–cell interaction prediction analysis
We conducted cell–cell interaction analysis by using CellphoneDB (v.5; https://github.com/ventolab/CellphoneDB) with method: statistical inference of interaction specificity on default setting. For each sample, we calculated interactions for CD4 TCM cells and core memory B cells. We retained the interactions with an adjusted P value below 0.05. For group comparisons, interactions with ≤15 counts in either age group were excluded.
TEA-seq analysis
TEA-seq datasets were directly downloaded from GEO (GSE214546). We performed label transfer, doublet detection and QC on the basis of the RNA module. Label transfer was conducted using Celltypist, on the basis of the models generated from our atlas reference. We filtered the cells if predicted labels were not T cells. Doublet detection was done using the scanpy.external.pp.scrublet function on the RNA module only. Predicted doublets were filtered out before any downstream processes. QC was performed by ensuring that pct_counts_mito was below 15%, and n_genes_by_counts was either below 2,500 or above 200. Cells from the RNA module were matched to those from the ATAC module using original barcodes and well IDs. Any mismatched cells containing only RNA or only ATAC modules were excluded. We performed scATAC-seq analysis on ArchR (v.1.0.2)64. The following steps were executed using the default ArchR workflow: LSI dimensionality reduction, group coverage, identification of reproducible peaks (MACS3)65, peak matrix construction, motif annotation (cisbp database), background peak construction, deviation matrix and weight imputation (MAGIC)66. The z-scored ChromVar motif matrix was extracted with imputed weights. The motif scores at the sample level were calculated on the basis of the mean score for each cell type within each sample.
Trajectory analysis
We performed trajectory analysis with Slingshot67. First, we computed UMAP embeddings and Leiden clusters on the basis of the RNA module. Then, using Slingshot settings (approx_points=200 and extend = ‘n’), we inferred pseudotime and extracted trajectories. To compare motifs between the two age groups along a unified pseudotime axis, we applied loess smoothing to each group’s data, extracted and aligned their curves, and calculated the difference between the smoothed curves. Finally, we generated a plot with shaded regions representing positive and negative differences, highlighting where one group’s motif expression exceeds the other’s.
Statistical tests
Unless otherwise specified, all Wilcoxon tests were conducted as two-sided test by default and all reported correlations are based on Spearman correlation.
Data visualization
The following tools are provided to facilitate data exploration and discovery.
Human Immune Health Atlas (https://apps.allenimmunology.org/aifi/resources/imm-health-atlas/): (1) AIFI Immune Health Atlas UMAP Explorer. Explore cell subsets and gene expression in a UMAP viewer to display an overview of the entire dataset (https://apps.allenimmunology.org/aifi/resources/imm-health-atlas/vis/umap). (2) AIFI Immune Health Atlas Gene Expression Reference. A quick reference for gene expression across cell types and age groups (https://apps.allenimmunology.org/aifi/resources/imm-health-atlas/vis/reference/). (3) AIFI Immune Health Atlas Clinical Data Explorer. Flexible visualization of clinical metadata and lab results collected from our Atlas donors (https://apps.allenimmunology.org/aifi/resources/imm-health-atlas/vis/clinical/).
Dynamics of Human Immune Health and Age Resource: (https://apps.allenimmunology.org/aifi/insights/dynamics-imm-health-age/): (1) Sound Life Longitudinal Dynamics Explorer. (1) Explore gene expression and cell-type frequency across the longitudinally sampled blood draws within each healthy subject of our longitudinal cohort (https://apps.allenimmunology.org/aifi/insights/dynamics-imm-health-age/vis/dynamics/). (2) Sound Life Differential Gene Explorer. Browse results of pairwise DEG tests for age, sex, CMV and influenza vaccination between our cohort samples (https://apps.allenimmunology.org/aifi/insights/dynamics-imm-health-age/vis/deg/). (3) Sound Life Clinical Data Explorer. Flexible visualization of clinical metadata and lab results collected from our cohort’s donors (https://apps.allenimmunology.org/aifi/insights/dynamics-imm-health-age/vis/clinical/). (4) Sound Life Major Immune Cell Subsets UMAP Explorer. Explore CD4 T cell, CD8 T cell, B cell, NK cell, myeloid cell and other subsets and their gene expression separately for each major immune cell compartment at higher resolution in the entire Sound Life scRNA-seq data (https://apps.allenimmunology.org/aifi/insights/dynamics-imm-health-age/vis/umap/).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
First Appeared on
Source link

