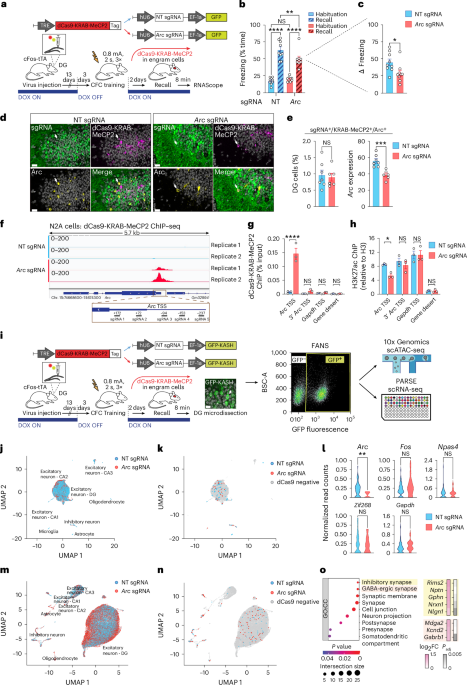
Cell-type- and locus-specific epigenetic editing of memory expression
All animals and procedures used in this study were approved by the Veterinary Office of the Federal Council of Switzerland under the animal experimentation license VD2808.2.
sgRNA design and cloning
To design guide sequences for the CRISPR–dCas9 systems a total of 439 bp spanning the promoter of the mouse gene Arc were used as input for the CRISPROR program38. Five sgRNAs were selected and DNA oligonucleotides corresponding to the sgRNA sequences (Supplementary Table 2) were cloned individually into pLenti SpBsmBI sgRNA Hygro (Addgene 62205) containing an U6-driven sgRNA scaffold. Afterwards, the individual sgRNAs were multiplexed using NEBridge Golden Gate Assembly kit (NEB) with custom designed primers and cloned into the lentiviral pLVX-EF1a-GFP backbone. The sgRNA sequence against the bacterial LacZ gene was used as an NT control sgRNA as previously described39. The plasmids for dCas9-VPR and dCas9-KRAB-MeCP2 were generated by subcloning the lenti-EF1a-dCas9-VPR-Puro (Addgene 99373) and the lenti-SYN-dCas9-KRAB-MeCP2 (gift from J. Day) into the lentiviral pLVX backbone under control of either the CMV promoter (in vitro experiments in N2A cells) or the TRE (in vivo experiments). The dCas9-CBP constructs were obtained by replacing the VPR part with the CBP catalytic domain we previously characterized40. For the AcrIIA4 experiments, the pLVX-DIO-hSyn-dCas9-VPR plasmid was generated by subcloning the dCas9-VPR sequence into the pLVX-DIO-hSyn backbone; the pLVX-hU6-Arc sgRNAs-EF1a-GFP-TRE-mCherry-P2A-AcrII4A (in vitro experiments) and the pLVX-TRE-mCherry-P2A-AcrII4A (in vivo experiments) were generated starting from pHR-Ef1a-mCherry-P2A-AcrIIA4 (Addgene 125148); the pSIN-hPGK-rtTA plasmid was a gift from D. Trono. For a full list of the plasmids used, see Supplementary Table 2.
Lentivirus production
Lentiviral vectors were produced as described previously41. HEK-293T cells (ATCC) were transfected using calcium phosphate with a second-generation packaging system composed of the pMD2G and psPAX2 vectors alongside the different pLVX vectors used in this study. After 4 days, medium was collected and centrifuged in an ultracentrifuge at 45,000g for 90 min at 4 °C. The pellet was resuspended in 0.5% bovine serum albumin (BSA)-PBS and viral titer determined using the HIV-1 p24 antigen ELISA kit (Zeptometrix Corp.). Injections were performed with 500 ng of each virus, which corresponded to a volume between 200 nl and 500 nl depending on the individual titer.
Cell culture and transfection
N2A cells (ATCC), were cultured in Dulbecco’s Modified Eagle Medium GlutaMAX (Gibco) supplemented with a 1% penicillin-streptomycin solution (Gibco) and 10% fetal bovine serum (Gibco). Cells were transfected with the appropriate plasmids using Lipofectamine 2000 (Invitrogen) according to manufacturer’s instructions. To induce the expression of the AcrIIA4 construct, cells were treated with 1 μg ml−1 DOX.
Immunocytochemistry
N2A cells were cultured on coverslips coated with poly-D-lysine (Corning) and transfected as described above. After 48 h, cells were fixed for 12 min at room temperature in a 4% paraformaldehyde (PFA), 4% sucrose PBS-based solution. Next, samples were incubated overnight at 4 °C with the primary antibody in a 0.3% Triton, 3% BSA, 10% serum PBS-based solution. The following day, cells were washed with PBS and incubated with the secondary antibody for 1 h at room temperature. Finally, coverslips were mounted on a slide with Vectashield–4′,6-diamidino-2-phenylindole (DAPI) and imaged with an Olympus Slide Scanner (Olympus VS120) at ×10 magnification. Antibodies used are listed in Supplementary Table 2.
Immunoprecipitation assay
N2A cells treated as described above were resuspended in immunoprecipitation buffer (20 mM Tris-HCl (pH 7.5), 100 mM NaCl, 1 mM NaCl, 1 mM EDTA, 0.1% Triton X-100), shortly sonicated (five cycles 3 s on–off, Bandelin Sonopuls sonicator) and centrifuged for 10 min at 16,000g at 4 °C. Following a 1:3 dilution in 10 mM Tris-HCl (pH 7.5), 150 mM NaCl and 0.5 mM EDTA, lysates were incubated at 4 °C with either Flag or CBP antibody for 2 h, and then with G Dynabeads overnight. The day after, G Dynabeads were washed three times with dilution buffer and eluted in Laemmli buffer. For western blots, protein lysates were resolved in 7.5% SDS–polyacrylamide gels and transferred to nitrocellulose membranes. Proteins were visualized with horseradish peroxidase-conjugated secondary antibodies and ECL detection reagents (Amersham). Antibodies used are listed in Supplementary Table 2.
RNA extraction and quantitative PCR
Total RNA was extracted using Trizol (Thermo Fisher Scientific), and cDNA synthesized using the SuperScript III First-Strand Synthesis kit (Invitrogen) according to the manufacturer’s instructions. All quantitative PCRs (qPCRs) were performed with the FAST SYBR Green Master Mix (Applied Biosystems). Each biological sample was loaded in technical triplicates and fluorescence acquisition carried out on a StepOnePlus Real-Time PCR System (Applied Biosystems). Calculations were performed using the ΔΔCt method and levels of Gapdh mRNA and Actin mRNA as reference genes. Primer sequences are listed in Supplementary Table 2.
Chromatin immunoprecipitation
Samples for chromatin immunoprecipitation (ChIP)–qPCR were obtained as described previously with minor adaptations42. N2A cells were crosslinked using 1% formaldehyde for 10 min at room temperature, followed by lysis in 5 mM HEPES (pH 8), 85 mM KCl, 0.5% NP-40. After centrifugation, nuclei were washed in 5 mM HEPES (pH 8), 85 mM KCl and then sonicated in 1 ml of 50 mM Tris-HCl (pH 8), 10 mM EDTA, 1% SDS (Covaris LE220 sonicator). The resulting chromatin was precleared with A or G Dynabeads (Invitrogen) and incubated overnight with antibodies in immunoprecipitation (IP) buffer (50 mM HEPES (pH 7.5), 1% Triton X-100, 150 mM NaCl). Next, A or G Dynabeads were added for 6 h, followed by eight washes: the first three in IP buffer plus 0.1% sodium deoxycholate, 0.1% SDS and 1 mM EDTA; the second three in the same buffer but with 500 mM NaCl; then one wash in 10 mM Tris-HCl pH 8, 250 mM LiCl, 1 mM EDTA, 1% NP-40 and 0.5% sodium deoxycholate; and a final wash in 10 mM Tris-HCl pH 8, 1 mM EDTA. Chromatin was then eluted in 1% SDS, 0.1 M NaHCO3 and incubated at 65 °C overnight for decrosslinking plus Proteinase K digestion, before clean up using the QiaQuick PCR purification kit (Qiagen). qPCR was carried out as for gene expression analysis. Values for each IP sample were normalized relative to corresponding input chromatin for the same treatment. Primer sequences and antibodies are listed in Supplementary Table 2.
ChIP–seq library preparation and analysis
Libraries from samples prepared as described above were generated using the NEBNext Ultra II DNA Library Preparation Kit (NEB) according to manufacturer’s instructions. Samples were multiplexed and 75-bp paired-end reads generated on a Nextseq500 (Illumina). After reads were trimmed for NEBNext Ultra II DNA (TruSeq) adapters, the FastQ files were demultiplexed using bclconvert (v.3.9.3, Illumina) and aligned to the mm10 genome using bowtie2 (v.2.4.5) in paired-end mode using default parameters. The R library Csaw (v.1.30.1) was used for peak calling, with peaks being considered nonsignificant for regions that had less than a threefold enrichment compared to their 2-kb neighborhood, and reads were normalized using the locally estimated scatterplot smoothing (loess) algorithm. Peaks were assigned to the nearest promoter with the distanceToNearest function from GenomicRanges, and a differential enrichment analysis performed using EdgeR (v.3.38.4). The threshold for significance was set at log fold change (FC) > 2 and P value < 0.05. When assigning peaks to genomic features promoter regions were defined as 2 kb upstream and 200 bp downstream of the gene transcription start site (TSS). ChIP–seq data were visualized using the Integrative Genomics Viewer (v.2.16.0). See Supplementary Table 1 for results summary.
Animals
cFos-tTA mice were bred inhouse from the original Jackson Laboratory (JAX strain 018306) on a C57Bl/6JR background. Animals were group-housed in a 12-h light/dark cycle with water and food available ad libitum. DOX (0.2 mg ml−1) was given orally through the water supply beginning at least 7 days before the start of the experiment, and only ceased 3 days before the session where the tagging window was desired. DOX was provided back to the animals as soon as the tagging window was no longer needed. Double transgenic cFos-CreERT2/R26-CAG-rtTALSL animals were generated inhouse by crossing the original JAX strains 030323 and 029617. cFos-CreERT2/R26-CAG-rtTALSL mice were injected with tamoxifen (4-OHT, Sigma-Aldrich, 50 mg kg−1) immediately after fear conditioning. Powdered tamoxifen was dissolved in ethanol 100% at a concentration of 20 mg ml−1 and stored at −20 °C. On the day of the experiment, tamoxifen was redissolved by shaking at 37 °C, then two volumes of corn oil were added and ethanol was evaporated shaking at 37 °C for a final concentration at 10 mg ml−1. Tamoxifen was kept at 37 °C until injection. Mice were between 8 weeks and 13 weeks old at the start of the experiments, and all were male. All behavioral procedures were performed between 1 pm and 5 pm local time and animals were assigned randomly to experimental groups.
Behavioral procedures
CFC experiments were performed using a TSE Multi Conditioning System. CFC encoding consisted of a first 3-min exploration phase, followed by 2-s long foot shocks spaced by 28 s. The number of repetitions of the pause-shock stage as well as the electrical current varied depending on the experiment (as illustrated in figures). After the last shock, the animal was left in the chamber for an additional 15 s and brought back to its home cage. The recall phase consisted of a 3-min exposure to the same context, without any shock. The time after encoding at which recall took place varied depending on the experiment. The movement of the animals was measured automatically using an infrared beam cut detection system (TSE Systems) and freezing calculated as the absence of movement for more than 500 ms. Open field experiments consisted of a 15-min long session during which the animals were left exploring an open arena (72 × 72 cm). Sessions were analyzed using Ethovision System (Noldus).
Virus injections
Surgeries were performed as described with minor adaptations43. Before surgery, mice were injected intraperitoneally with an anesthetic mix of fentanyl (0.05 mg kg−1), midazolam (5 mg kg−1), and metedomidin (0.5 mg kg−1), followed by a subcutaneous injection of an analgesic mix (lidocaine 6 mg kg−1 and bupivacaine 2.5 mg kg−1) at the surgery site. Animals were then placed on a stereotaxic micromanipulator frame (Kopf Instruments), skin disinfected with betadine and opened with a scalpel. To target the DG, holes were drilled in the skull with a 30-gauge drill bit at ±1.3 mm medio-lateral, −2.0 mm anterior–posterior. A virus-loaded micropipette (BLAUBRAND, intraMARK, tip diameter 10–20 µm) was lowered to −2.0 mm and 500 ng of viral particles were injected bilaterally at a rate of 0.2 µl min−1. After 5 min, the micropipette was raised 20 µm from the target for 1 min to prevent backflow and allow diffusion, then removed at 10 µm s−1. After the injections, the skin was sutured and atipamezol (2.5 mg kg−1) administered intraperitoneally for anesthesia reversal. Postsurgery, mice were moved to a clean cage on a heated pad until regaining mobility, then returned to their home cage with paracetamol provided in the drinking water for 1 week (500 mg per 250 ml). Surgery efficiency was verified by visualizing the GFP signal and misinjected brains were excluded from further analyses.
Nuclei preparation for scATAC-seq and single-cell RNA sequencing
Mice were euthanized 8 min after the recall session and the DG microdissected, flash-frozen and stored at −80 °C. Nuclei were extracted as described previously with minor adaptations44. To obtain a sufficient number of nuclei for scATAC-seq, the DGs from three animals were pulled for each condition. For single-cell RNA sequencing (scRNA-seq) each mouse was instead processed individually. All steps were performed at 4 °C. Brain tissue was first homogenized in homogenization buffer (250 mM sucrose, 25 mM KCl, 5 mM MgCl2, 10 mM Tris-HCl, pH 7.4, 1 mM dithiothreitol, 1× protease inhibitor, 1 U μl−1 RiboLock RNase inhibitor, 0.1% NP-40, in H2O), filtered through a 40-μm cell strainer and washed with nuclei isolation medium (without NP-40). Samples were centrifuged for 8 min at 1,000g, and nuclei resuspended in PBS/BSA storage solution (PBS 1×, 5 mM MgCl2, 0.5% BSA, 1 mM dithiothreitol in H2O) followed by filtering through 35-μm strainers. FANS was performed with a Sony SH800 sorter using a 100-μm nozzle and nuclei sorted on the basis of the GFP signal. For scATAC-seq, sorted nuclei were incubated for 2 min in permeabilization buffer (nuclei isolation medium plus 0.01% Tween, 0.001% Digitonin, 0.01% NP-40, 0.5% BSA), then washed in nuclei isolation buffer plus 0.5% BSA, resuspended in diluted nuclei buffer (Chromium nuclei isolation kit, 10x Genomics) and finally processed for GEMs generation using Chromium X and the ATAC NextGEM v.2 kit (10x Genomics). For scRNA-seq, sorted nuclei were fixed using the Evercode Low Input Fixation kit (Parse Biosciences) according to the manufacturer’s instructions and stored at −80 °C until libraries preparation.
scATAC-seq and scRNA-seq library preparation and analyses
The individual scATAC-seq libraries matching each condition (Arc or NT sgRNA) were pooled and 65-bp paired-end reads were generated on an Aviti platform (Element Biosciences), with a mean depth of ~200,000 reads per cell. Raw files were aligned and features were counted using default parameters for Cellranger-atac count45. The R tools Signac46 and Seurat47 with default parameters were used for downstream processing. Cells were maintained if they had between 300 and 75,000 unique molecular identifier (UMI) counts and genes were kept if they had greater than 30 reads, a TSS enrichment of more than two and a nuclear signal less than four. Cell types were defined by using the values calculated in the gene activity matrix with MapMyCells48. Cas9-positive cells were defined by performing a targeted PCR on each library, sequencing and then searching sequences matching the 3’ end of the Cas9 gene and extracting the cell barcode. Cells were considered Cas9 positive if the barcode has at least one Cas9 sequence hit in the PCR analysis or the initial library sequencing run. For the dCas9-positive cells across experimental conditions the normalized reads count at the TSS location of genes of interest were extracted and plotted as violin plots. For scRNA-seq, individual libraries from four Arc sgRNA and five NT sgRNA mice were prepared using the Evercode WT v.3 kit (Parse Biosciences) before being combined into eight sublibraries according to the manufacturer’s instructions. Sublibraries were sequenced on an Aviti platform (Element Biosciences) with a reads configuration of 150-bp paired-end and at depth of ~100 million reads per sample. Next, raw files were aligned with Parse Biosciences Trailmaker (v.1.4.1) to a modified version of the mouse genome (mm10) that includes GFP and Cas9m4 sequences. Seurat47 was used to filter for cells that had between 300 and 10,000 expressed genes, between 500 and 100,000 UMI counts and fewer than 1% mitochondrial reads. Note, only genes that were present in at least 20 cells were considered for further analysis. Each library was then separately normalized using SCTransform49 on the top 3,000 features before all libraries were integrated together. MapMyCells48 was used to identify cell types and Cas9-positive cells were defined as being any cells with at least 1 UMI count in the Cas9 sequence region. Finally, the Seurat47 command FindMarkers() was used to define differentially expressed genes between cells coming from Arc and NT samples in each cell type, with a P value ≤ 0.01 as cut-off. A summary of the scATAC-seq and scRNA-seq results is provided in Supplementary Table 1.
Histology and RNAscope
Eight minutes after the last behavioral test, animals were anesthetized with pentobarbital (150 mg kg−1) and perfused transcardially with first PBS and then 4% PFA in PBS. Brains were extracted, postfixed overnight in 4% PFA, transferred in a cryoprotectant solution (30% sucrose in PBS) for at least 48 h, and frozen at −80 °C. Sections of 20 μm were cut using a cryostat, and stored at −20 °C in an antifreeze solution (30% ethylene glycol, 15% sucrose, 0.02% azide in PBS). RNAscope followed by immunohistochemistry was performed using the RNAscope Multiplex Fluorescent v.2 kit (ACD Bio) according to the manufacturer’s instructions. Probes of interest (Supplementary Table 2) were hybridized for 2 h at 40 °C and the signal was amplified through AMP1, AMP2 and AMP3 probes before being developed using the appropriate horseradish peroxidase signal. Finally, the presence of GFP was revealed using a standard immunohistochemistry protocol. Slices were blocked 30 min in 1% BSA-PBS and incubated overnight at 4 °C with the anti-GFP primary antibody (Supplementary Table 2) in 1% BSA-PBS. The next day, the secondary antibody (Supplementary Table 2) was applied for 1 h at room temperature and nuclei were stained with Hoechst dye. The immunohistochemistry experiment of Extended Data Fig. 1 was performed as described previously43 using the primary and secondary antibodies listed in Supplementary Table 2.
Confocal images acquisition and analysis
An Upright Leica DM6 CS laser scanning confocal microscope was used to acquire images of the DG for each brain slice. All slides were acquired at ×63 magnification with a resolution of 512 × 512, speed of 400 Hz, airy unit between 0.3 and 0.4 a.u. and line averaging of three. Channels were acquired from the longest wavelength to the shortest wavelength and acquired one at a time to avoid leakage between the channels with spectrum overlaps assessed with BD spectrum viewer (BD Biosciences). A tilescan of the acquired area was then stitched within the Leica LAS-X software. Images were analyzed using QuPath (v.0.2.3 to v.0.4.3)50. For each confocal image of a single DG, detected nuclei were first classified as part of the DG granular cell layer or not using a machine-learning based algorithm developed inhouse starting from the StarDist library. Next, nuclei were defined as positive for each given channel if their mean signal was higher than a manually preset threshold of detection. Finally, measurements from all detected nuclei were exported and analyzed using Phyton scripts. To compute Arc expression levels for the triple-positive nuclei (sgRNA+, dCas9+, Arc+), the mean intensity value corresponding to the Arc probe signal for each nucleus in a given DG was extracted and averaged to obtain a single value per acquired image. Overall, a total of four images was analyzed for each mouse, and three to four mice per group were compared in each experiment.
Statistics and reproducibility
Statistical analysis was performed in Prism 9 (GraphPad) and all data are displayed as mean ± s.e.m. For in vivo experiments, no statistical methods were used to predetermine sample sizes, but the number of animals used in each experiment is similar to those reported in previously published engram studies. Data distribution was assumed to be normal, although this was not formally tested. Animals were assigned randomly to experimental groups and age matched. For in vitro experiments, at least three independent experiments were performed for statistical analysis unless otherwise specified in the figure legends. The statistical test used and the definition of n are described in the figure legends. Statistical analysis details for each figure are reported in Supplementary Table 3.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
First Appeared on
Source link

